



Transkript
FORTBILDUNG
Krankheitsmodifizierende Therapie des Morbus Parkinson
Die Entwicklung einer potenten neuroprotektiven Therapie zählt zu den wichtigsten Zielen der Parkinson-Forschung. Die vergangenen Jahre haben nicht eindeutige Hinweise auf ein krankheitsmodifizierendes Potenzial von Dopamin-Agonisten und MAO-B-Inhibitoren erbracht. Verbesserungen im Studienprotokoll zur sensitiven Erfassung von klinischen Unterschieden und in den biologischen Massen des Krankheitsfortschritts können in Zukunft helfen, positive Effekte klarer zu identifizieren. Vor allem sind aber offensichtlich neue Medikamente mit potenterem Wirkmechanismus nötig. Aktuell befinden sich biologisch sehr unterschiedliche Interventionen in klinischer Testung, die unser aktuelles pathophysiologisches Konzept der Parkinson-Krankheit von verschiedenen Seiten her testen.
Sylvia Maass Günter Höglinger
3/2013
von Sylvia Maass und Günter Höglinger
Einleitung
D ie vergangenen Jahre haben nicht eindeutige Hinweise auf ein krankheitsmodifizierendes Potenzial von Dopaminagonisten und MAO-B-Inhibitoren erbracht. Verbesserungen im Studienprotokoll zur sensitiven Erfassung von klinischen Unterschieden und in den biologischen Massen des Krankheitsfortschritts können in Zukunft helfen, positive Effekte klarer zu identifizieren. Ebenso bedarf das Konzept von sogenannten Futility-Trials (3, klinischen Machbarkeitsstudien) einer weiteren Verfeinerung zur Selektion von Medikamenten aus der präklinischen Entwicklung für grosse Effizienzstudien. Vor allem sind aber offensichtlich neue Medikamente mit potenterem Wirkmechanismus nötig. Aktuell befinden sich biologisch sehr unterschiedliche Interventionen in klinischer Testung (siehe Kasten), die unser aktuelles pathophysiologisches Konzept der Parkinson-Krankheit von verschiedenen Seiten her testen. Hierzu zählen unter anderem eine alpha-Synuclein-Vaccine, Nicotin, eine anti-aggregatorische Substanz (EGCG), eine Neurotrophin-artige Substanz (Cogane), eine Urat-steigernde Substanz (Inosin), ein Calciumkanalblocker (Isradipin), ein Radikalfänger (N-Acetylcystein), bioenergetische (Creatin, Coenzym Q10) und antiinflammatorische Substanzen (Pioglitazon, AZD3241). Neben einer Klärung der pathophysiologischen Relevanz dieser Mechanismen lassen sich aus diesen Studien hoffentlich auch die dringend benötigten neuen therapeutischen Wirkprinzipien ableiten.
Dieser Artikel stellt die klinische Relevanz vorhandener Ergebnisse dar und gibt einen umfassenden Überblick über aktuelle Entwicklungen.
Neuroprotektion beim M. Parkinson Trotz intensiver Forschungstätigkeit in den vergangenen Jahrzehnten stehen derzeit noch keine gesicherten neuroprotektiven Therapieansätze für den klinischen Einsatz zur Verfügung. Die Entwicklung neuroprotektiver beziehungsweise krankheitsmodifizierender Therapien wird kompliziert durch die Tatsache, dass die Manifestation klinisch fassbarer Symptome zu einem Zeitpunkt beginnt, an dem nur mehr 30 Prozent striatales Dopamin verfügbar ist. Das sind geschätzt zirka 20 Jahre nachdem der neurodegenerative Prozess überhaupt begonnen hat. Eine neuroprotektive Behandlung, die eine Verlangsamung des Zellverlustes zum Ziel hat, sollte jedoch sinnvollerweise vor Beginn der klinisch manifesten Phase begonnen werden, in der auch typischerweise erstmals die symptomatische Therapie mittels Dopamin-Substitution erfolgt. Alle aktuell angewandten neuroprotektiven Ansätze kommen aber erst im klinisch manifesten Stadium der Erkrankung zum Tragen – in einer Phase also, in der zu diskutieren ist, ob der neuroprotektive Ansatz nicht zu spät erfolgt.
Evidenz für krankheitsmodifizierende Therapieansätze 1. Coenzym Q10 als Therapie gegen mitochondriale Dysfunktion: In experimentellen Vorarbeiten (4) hatte sich eine reduzierte Menge an Coenzym Q10 (CoQ10) in den Mitochondrien von Parkinson-Patienten sowie eine gestörte Funktion der Mitochondrien, den «Kraftwerken» der Zellen, gefunden. Letztere resultiert in einer verminderten Aktivität von Komplex-I der Atmungskette, was in der Folge zu einer Störung des Elektronentransports, Defekten der oxidativen Phosphorylierung, zellulärem ATP-Verlust und vermehrter Bildung freier Radikale und oxidativem Stress führt. CoQ10 ist der physio-
&PSYCHIATRIE NEUROLOGIE
21
FORTBILDUNG
logische Elektronenempfänger von Komplex-I und vermittelt den Elektronentransport von Komplex-I zu Komplex-III. Ferner kann es freie Radikale abfangen und die Toxizität von MPTP (1-methyl-4-phenyl-1,2,3,6tetrahydropyridine) und Rotenone im Tierversuch reduzieren (5). Die Rationale der Studien, welche CoQ10 bei Patienten mit M. Parkinson supplementieren, ist die erwiesene Verbesserung der mitochondrialen Funktion durch oral zugeführtes CoQ10. In einer kalifornischen klinischen Studie (1) wurden 80 Parkinson-Patienten randomisiert, die entweder 300, 600 oder 1200 mg CoQ10 pro Tag oder Plazebo über 16 Monate erhielten. In viermonatigen Abständen erfolgte eine standardisierte neurologische Untersuchung. Hier zeigte sich ein Trend zu einer reduzierten körperlichen Beeinträchtigung (gemessen mittels Unified Parkinson´s Disease Rating Scale [UPDRS]) in der Gruppe der Patienten mit 1200 mg/Tag im Vergleich zur Plazebogruppe. Eine in Deutschland durchgeführte multizentrische Studie (2) (Storch et al, 2007) untersuchte 130 Parkinson-Patienten ohne Wirkungsfluktuationen mit stabilem Behandlungsregime. Dabei wurden 300 mg CoQ10 versus Plazebo über 3 Monate verabreicht und monatlich der UPDRS dokumentiert. Es zeigte sich, dass durch Einnahme von 100 mg CoQ10 dreimal täglich ähnliche Plasmaspiegel erreicht werden konnten wie bei den Patienten aus der amerikanischen Studie. Es fand sich jedoch kein signifikanter Unterschied zwischen den beiden Gruppen hinsichtlich der motorischen Beeinträchtigung gemessen am UPDRS. Somit konnten keine Hinweise auf symptomatische Effekte von CoQ10 bei Patienten im mittleren Krankheitsstadium gefunden werden. Zusammenfassend lässt sich feststellen, dass CoEnzym Q10 in der Behandlung des M. Parkinson offensichtlich nicht symptomatisch wirksam ist, jedoch möglicherweise protektiv im Sinne einer Krankheitsverzögerung wirkt, wobei hierfür der endgültige Beweis noch zu erbringen ist.
2. MAO-B-Hemmer als antioxidative Therapie Aufgrund der Tatsache, dass MAO-B-Hemmer im Experiment die Bildung freier Radikale vermindern sowie den Effekt des dopaminergen Neurotoxins MPTP im Tiermodell blockieren, stellte sich die Frage, ob die Behandlung mit einem MAO-B-Hemmer einen neuroprotektiven Effekt bei Patienten mit M. Parkinson erzielen kann.
2.1 Selegilin: Die DATATOP-Studie (6) war eine plazebokontrollierte klinische Studie, die die Hypothese untersuchte, ob eine langfristige Therapie mit Deprenyl 10 mg/Tag und/oder Tocopherol (Vitamin E) 2000 IU/Tag bei Patienten im frühen Stadium der Parkinson-Erkrankung den Zeitpunkt für eine Therapie mit L-DOPA herauszögern kann (primärer Endpunkt). In 28 nordamerikanischen Zentren wurden 800 geeignete De-novo-Patienten in den frühen Stadien der Erkrankung eingeschlossen. Sie erhielten entweder Selegilin, Vitamin E, Selegilin und Vitamin E in der Kombination oder Plazebo. Die Teilnehmer wurden alle 3 Monate systematisch über einen Zeitraum von 2 Jahren evaluiert. Dabei wurde bestimmt, ob und wann der primäre Endpunkt, die Notwendigkeit des Beginns einer L-DOPA-Therapie, er-
reicht wurde. Bei der Baseline-Untersuchung bestand die Kohorte nur aus minimal beeinträchtigten Patienten, die keine symptomatische Anti-Parkinson-Medikation benötigten. In den Ergebnissen zeigte sich, dass für Patienten, die mit Selegilin behandelt worden waren, die Notwendigkeit einer L-DOPA-Therapie durchschnittlich um 9 Monate herausgezögert werden konnte, im Vergleich zu plazebobehandelten Patienten. Allerdings gab es an der Studie beziehungsweise dem Studiendesign einige Kritikpunkte (7): 1. Der primäre Endpunkt, das heisst, das Vorliegen einer
Beeinträchtigung, die eine L-DOPA-Therapie erfordert, basierte auf den subjektiven Bedürfnissen der Patienten und nicht auf objektivierbaren Messwerten (wie z.B. motorische Geschwindigkeit). Insofern ist eine valide Krankheitsprogression nicht sicher gegeben. 2. Die Follow-up-Periode war zu kurz, da der durchschnittliche Zeitraum bis zum Erreichen des Endpunkts bei 9 Monaten lag. Als adäquate Follow-upPeriode sollte die 2-Jahres-Periode gelten, die als Beobachtungszeitraum der Studie veranschlagt wurde. 3. Evidenz für einen direkten symptomatischen Therapie-Effekt: Die klinische Verbesserung der behandelten Patienten setzte sehr früh ein (signifikant innerhalb der ersten 3 Monate). Nach Absetzen von Selegilin zeigte sich nach einem Monat kein Washout-Effekt, zwei Monate nach Absetzen von Selegilin zeigte sich jedoch eine signifikante Verschlechterung, was für einen symptomatischen Effekt der Therapie mit Selegilin spricht. Diese Kritikpunkte legen nahe, dass der beobachtete positive Effekt im Wesentlichen aus einem symptomatischen, aber möglicherweise nicht neuroprotektiven Effekt von Selegilin resultierte.
2.2 Rasagilin: Bei der Adagio-Studie (Attenuation of Disease Progression with Azilect Given Once daily [8]) handelte es sich um eine doppelblinde Studie mit dem MAO-B-Hemmer Rasagilin, wobei insgesamt 1176 De-Novo-Parkinson-Patienten eingeschlossen wurden. Rasagilin wurde in der sogenannten «Early-Start»-Gruppe in einer Dosierung von entweder 1 mg oder 2 mg/Tag über 72 Wochen verabreicht. Die «Delayed-start»-Gruppe erhielt Plazebo über 36 Wochen und anschliessend über 36 Wochen Rasagilin in einer Dosis von entweder 1 mg oder 2 mg pro Tag. Die frühe Behandlung mit Rasagilin in einer Dosis von 1mg/Tag zeigte in der Tat eine motorische Verbesserung, die mit einem möglichen krankheitsmodifizierenden Effekt konsistent war. Diese Ergebnisse waren in der Gruppe der Patienten mit früher Behandlung mit 2 mg/Tag Rasagilin jedoch nicht nachweisbar. Aus verschiedenen Gründen müssen die Ergebnisse mit Vorsicht interpretiert werden. Schwer nachzuvollziehen ist der Unterschied im funktionellen Ergebnis zwischen der 1 mg- und 2 mg-Rasagilin-Gruppe; es ist schwer erklärbar, dass ein protektiver Effekt durch Verdopplung der Dosis verloren gehen würde. Es wurde auch die Frage gestellt, ob ein ausgeprägter symptomatischer Effekt der 2 mg-Dosis einen Benefit verschleiern kann bei Patienten der «Early-start»-Gruppe, die nur eine sehr milde Symptomatik aufwiesen. Dafür spricht eine Post-hoc-Subgruppen-Analyse, die einen
&22 3/2013 PSYCHIATRIE NEUROLOGIE
FORTBILDUNG
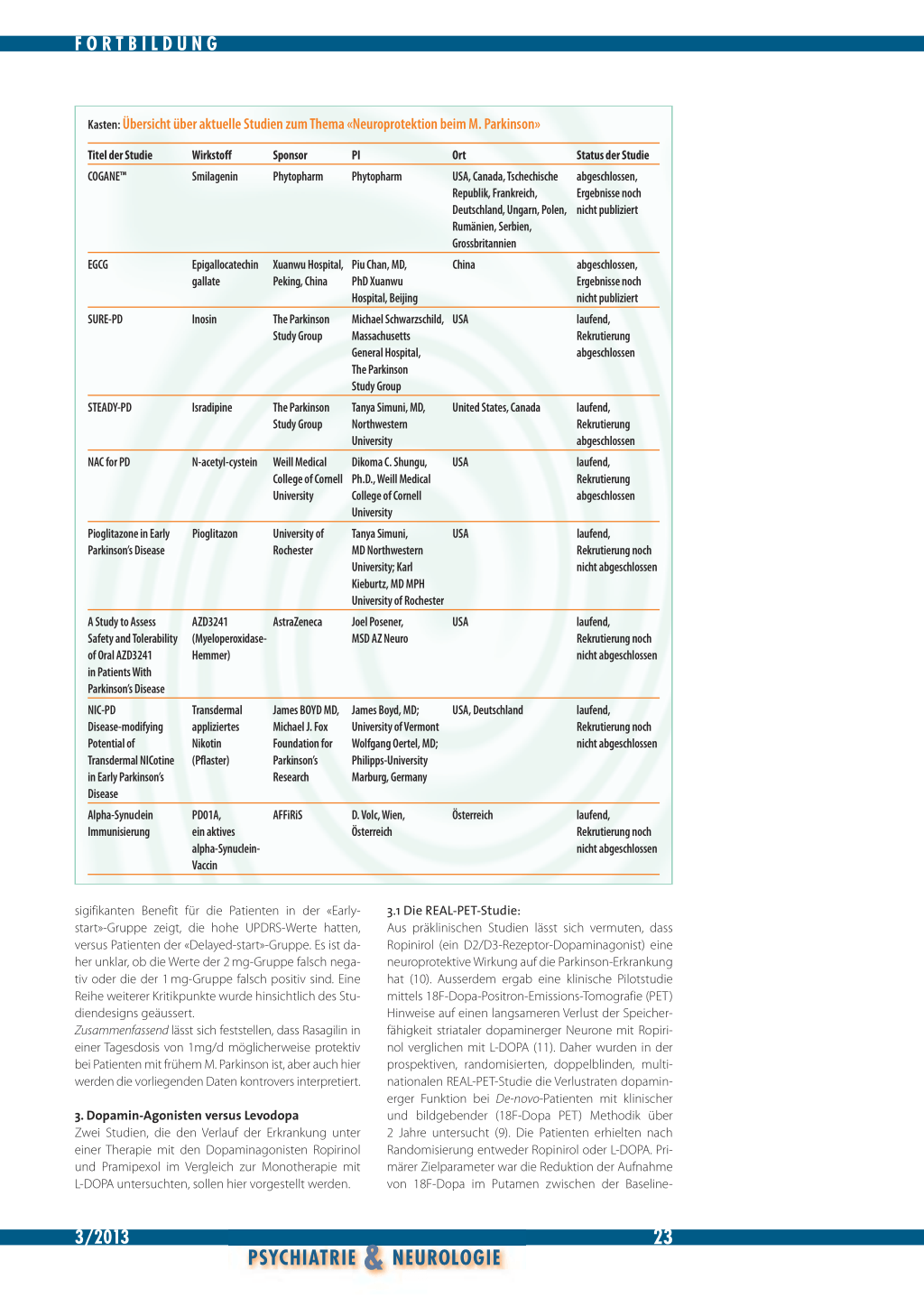
Kasten: Übersicht über aktuelle Studien zum Thema «Neuroprotektion beim M. Parkinson»
Titel der Studie COGANE™
EGCG
SURE-PD
STEADY-PD
NAC for PD
Pioglitazone in Early Parkinson’s Disease
A Study to Assess Safety and Tolerability of Oral AZD3241 in Patients With Parkinson’s Disease NIC-PD Disease-modifying Potential of Transdermal NICotine in Early Parkinson’s Disease Alpha-Synuclein Immunisierung
Wirkstoff
Sponsor
PI
Ort
Status der Studie
Smilagenin
Phytopharm Phytopharm
USA, Canada, Tschechische abgeschlossen,
Republik, Frankreich,
Ergebnisse noch
Deutschland, Ungarn, Polen, nicht publiziert
Rumänien, Serbien,
Grossbritannien
Epigallocatechin Xuanwu Hospital, Piu Chan, MD,
gallate
Peking, China PhD Xuanwu
Hospital, Beijing
China
abgeschlossen, Ergebnisse noch nicht publiziert
Inosin The Parkinson Michael Schwarzschild, USA Study Group Massachusetts General Hospital, The Parkinson Study Group
laufend, Rekrutierung abgeschlossen
Isradipine
The Parkinson Study Group
Tanya Simuni, MD, Northwestern University
United States, Canada
laufend, Rekrutierung abgeschlossen
N-acetyl-cystein
Weill Medical Dikoma C. Shungu,
College of Cornell Ph.D., Weill Medical
University
College of Cornell
University
USA
laufend, Rekrutierung abgeschlossen
Pioglitazon
University of Rochester
Tanya Simuni,
USA
MD Northwestern
University; Karl
Kieburtz, MD MPH
University of Rochester
laufend, Rekrutierung noch nicht abgeschlossen
AZD3241
AstraZeneca
(Myeloperoxidase-
Hemmer)
Joel Posener, MSD AZ Neuro
USA
laufend, Rekrutierung noch nicht abgeschlossen
Transdermal appliziertes Nikotin (Pflaster)
James BOYD MD, Michael J. Fox Foundation for Parkinson’s Research
James Boyd, MD; University of Vermont Wolfgang Oertel, MD; Philipps-University Marburg, Germany
USA, Deutschland
PD01A, ein aktives alpha-SynucleinVaccin
AFFiRiS
D. Volc, Wien, Österreich
Österreich
laufend, Rekrutierung noch nicht abgeschlossen
laufend, Rekrutierung noch nicht abgeschlossen
sigifikanten Benefit für die Patienten in der «Earlystart»-Gruppe zeigt, die hohe UPDRS-Werte hatten, versus Patienten der «Delayed-start»-Gruppe. Es ist daher unklar, ob die Werte der 2 mg-Gruppe falsch negativ oder die der 1 mg-Gruppe falsch positiv sind. Eine Reihe weiterer Kritikpunkte wurde hinsichtlich des Studiendesigns geäussert. Zusammenfassend lässt sich feststellen, dass Rasagilin in einer Tagesdosis von 1mg/d möglicherweise protektiv bei Patienten mit frühem M. Parkinson ist, aber auch hier werden die vorliegenden Daten kontrovers interpretiert.
3. Dopamin-Agonisten versus Levodopa Zwei Studien, die den Verlauf der Erkrankung unter einer Therapie mit den Dopaminagonisten Ropirinol und Pramipexol im Vergleich zur Monotherapie mit L-DOPA untersuchten, sollen hier vorgestellt werden.
3.1 Die REAL-PET-Studie: Aus präklinischen Studien lässt sich vermuten, dass Ropinirol (ein D2/D3-Rezeptor-Dopaminagonist) eine neuroprotektive Wirkung auf die Parkinson-Erkrankung hat (10). Ausserdem ergab eine klinische Pilotstudie mittels 18F-Dopa-Positron-Emissions-Tomografie (PET) Hinweise auf einen langsameren Verlust der Speicherfähigkeit striataler dopaminerger Neurone mit Ropirinol verglichen mit L-DOPA (11). Daher wurden in der prospektiven, randomisierten, doppelblinden, multinationalen REAL-PET-Studie die Verlustraten dopaminerger Funktion bei De-novo-Patienten mit klinischer und bildgebender (18F-Dopa PET) Methodik über 2 Jahre untersucht (9). Die Patienten erhielten nach Randomisierung entweder Ropinirol oder L-DOPA. Primärer Zielparameter war die Reduktion der Aufnahme von 18F-Dopa im Putamen zwischen der Baseline-
3/2013
&PSYCHIATRIE NEUROLOGIE
23
FORTBILDUNG
Visite und nach 2 Jahren. Es konnten 162 randomisierte Patienten ausgewertet werden. Eine verblindete, zentral durchgeführte Region-of-interest-Analyse zeigte eine signifikant geringere Reduktion im Putamen über 2 Jahre unter einer Therapie mit Ropinirol (13,4%; n = 68) verglichen mit L-DOPA (20,3%; n = 59).
3.2 Die CALM-PD-Studie: Die CALM-PD-Studie (12) untersuchte ähnlich wie die REAL-PET-Studie (9) mittels nuklearmedizinischer Bildgebung (DatScan-SPECT) das Voranschreiten des M. Parkinson unter einer Therapie mit dem Dopaminagonisten Pramipexol im Vergleich zu einer Monotherapie mit Levodopa zur Frage des Voranschreitens unter Pramipexol vs. Levodopa. Es wurden 82 Patienten in 17 nordamerikanischen Zentren zwischen 1996 und 1997 im Frühstadium der Erkrankung eingeschlossen, die dopaminerge Therapie benötigten, um aufkommende körperliche Einschränkungen zu behandeln. Nach Randomisierung erhielten die Patienten entweder Pramipexol 3 x 0,5 mg/d mit einem L-DOPA-Plazebo (n = 42) oder dreimal täglich Levodopa/Carbidopa 100/25 mg mit einem Pramipexol-Plazebo (n = 40). Für Patienten mit darüber hinaus bestehenden körperlichen Einschränkungen wurde die Dosis während der ersten 10 Wochen eskaliert, und in der Folge konnte L-DOPA open-label hinzugezogen werden. Nach 24 Monaten Follow-up konnte die Dosierung der Studienmedikation weiter modifiziert werden. Die primäre OutcomeVariable war die prozentuale Veränderung der striatalen [123I]-CIT-Aufnahme nach 46 Monaten im Vergleich zum Ausgangswert. Die prozentualen und absoluten Veränderungen der Aufnahme von [123I]-CIT im Striatum, Putamen und Ncl. caudatus nach 22 und 34 Monaten wurden ebenfalls untersucht. Der klinische Schweregrad der Erkrankung wurde mittels UPDRS 12 Stunden nach der letzten Anti-Parkinson-Medikamenten-Einnahme ermittelt. Es fand sich ein fortschreitender Verlust des präsynaptischen Dopamintransporters bei allen Parkinson-Patienten, jedoch fanden sich signifikant niedrigere Verlustraten bei Patienten, die mit Pramipexol behandelt waren, im Vergleich zu Patienten unter einer L-DOPA-Therapie. Nachdem [123I]betaCIT-SPECT als ein quantitativer Biomarker für striatale dopaminerge Nervenendigungen gelten kann, lässt sich aus diesen Daten ableiten, dass eine Behandlung mit Pramipexol im Vergleich zu Levodopa die Neurodegeneration der Parkinson-Erkrankung modifiziert. Zusammenfassend legen die bildgebenden Befunde sowohl der REAL-PET als auch der CALM-PD-Studie nahe, dass die Dopaminagonisten Ropinirol und Pramipexol im Vergleich zu Levodopa im frühen Stadium den Krankheitsverlauf günstig beeinflussen können. Einschränkend muss aber gesagt werden, dass bei beiden Studien keine eindeutigen klinischen Korrelate im Sinne einer Verbesserung der Motorik der Patienten gefunden wurde. Die klinische Relevanz der bildgebenden Befunde ist also höchst unklar.
Ausblick Abschliessend sei betont, dass die vergangenen Jahre aus der epidemiologischen, genetischen, neuropathologischen und experimentellen Forschung enorme neue Einsichten in die Pathophysiologie des M. Parkin-
Merkpunkte:
● Eine Studie (1), die Coenzym Q 10 bei Patienten mit M. Parkinson supplementierte, zeigte Hinweise für neuroprotektive Effekte; eine Bestätigung der Studienergebnisse steht aus.
● Studien mit selektiven MAO-B-Hemmern zeigten uneindeutige Ergebnisse: während Selegilin einen symptomatischen Effekt, jedoch vermutlich keinen krankheitsmodifizierenden Effekt zeigte (6, 7), kann Rasagilin in einer Tagesdosis von 1 mg möglicherweise einen krankheitsmodifizierenden Effekt haben (8).
● Bildgebende Studien legen neuroprotektive Effekte für die Dopaminagonisten Ropinirol (9, 10, 11) und Pramipexol (12) im Vergleich zu Levodopa nahe, deren klinische Relevanz ist aber nicht erwiesen.
● Aktuelle Studien untersuchen die neuroprotektiven Effekte von verschiedenen Substanzen(Smilagenin, Grüntee-Polyphenole, Inosin, Calciumantagonisten, N-Acetyl-Cystein, Nicotin, alpha-Synuclein-Vaccine sowie orale Antidiabetika und Myeloperoxidasehemmer [13–20]).
son generiert haben. Daraus wurden viele neue thera-
peutische Konzepte abgeleitet, die sich derzeit in der
klinischen Erprobung befinden. Die Frage nach Neuro-
protektion bei M. Parkinson wird derzeit im Rahmen
zahlreicher klinischer Studien untersucht (13–20). Einen
Überblick über die derzeit verfolgten Konzepte gibt
nachfolgender Kasten, der keinen Anspruch auf voll-
ständige Abbildung der aktuellen Forschungsland-
schaft erhebt. Zusammenfassend sei betont, dass die
klinische Erforschung neuroprotektiver Therapiean-
sätze für den M. Parkinson aus den vergangenen klini-
schen Studien sehr viele Erkenntnisse produziert hat
und auf der Basis dieses Wissens hoffentlich in naher
Zukunft durchgreifendere Erfolgsmeldungen zu ver-
melden sind.
●
Korrespondenzadresse:
Prof. Dr. med. Günter U. Höglinger
Dr. med. Sylvia Maass
Technische Universität München
Klinik für Neurologie
Abt. für Translationale Neurodegeneration
Max Lebsche Platz 30
D-81377 München (D)
E-Mail: Guenter.Hoeglinger@dzne.de
Anhang: - Conflict of Interest Prof. Dr. Höglinger erhielt in der Vergangenheit Honoraria für Advisory-Board-Arbeit, Vorträge oder Forschungskooperationen von Boehringer Ingelheim, Bristol-Myers Squibb, Elan, GlaxoSmithKline, Noscira, Proteosys, Roche, Teva, UCB.
&24 3/2013 PSYCHIATRIE NEUROLOGIE
FORTBILDUNG
Literaturverzeichnis:
1. Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perlmutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M; Parkinson Study Group. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002 Oct; 59(10): 1541–50.
2. Storch A, Jost WH, Vieregge P, Spiegel J, Greulich W, Durner J, Müller T, Kupsch A, Henningsen H, Oertel WH, Fuchs G, Kuhn W, Niklowitz P, Koch R, Herting B, Reichmann H; German Coenzyme Q(10) Study Group. Randomized, double-blind, placebo-controlled trial on symptomatic effects of coenzyme Q (10) in Parkinson disease. ArchNeurol. 2007 Jul; 64(7): 938–44.
3. NINDSNET-PD Investigators. A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology. 2007 Jan 2; 68(1): 20–8.
4. Shults CW, Haas RH, Passov D, Beal MF.: Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann Neurol. 1997 Aug; 42(2): 261–4.
5. Beal MF, Matthews RT, Tieleman A, Shults CW.: Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998 Feb 2; 783(1): 109–14.
6. Parkinson Study Group. The effect of selegiline on the progression of disability in early Parkinson´s disease. N Engl J Med 1989; 321: 1364–71.
7. Ward CD.: Does selegiline delay progression of Parkinson´s disease? A critical re-evaluation of the DATATOP study. J Neurol Neurosurg Psychiatry 1994; 57: 217–220.
8. Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, Langston W, Melamed E, Poewe W, Stocchi F, Tolosa E.: ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med. 2009 Sep 24; 361(13): 1268–78.
9. Whone AL, Watts RL, Stoessl AJ, Davis M, Reske S, Nahmias C, Lang AE, Rascol O, Ribeiro MJ, Remy P, Poewe WH, Hauser RA, Brooks DJ.: REAL-PET Study Group. Slower Progression of Parkinson’s Disease with Ropinirole versus Levodopa: The REAL-PET Study. Ann Neurol. 2003 Jul; 54(1): 93–101.
10. Iida M, Miyazaki I, Tanaka K, Kabuto H, Iwata-Ichikawa E, Ogawa N.: Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res. 1999 Aug 14; 838(1–2): 51–9.
11. Rakshi JS, Pavese N, Uema T, Ito K, Morrish PK, Bailey DL, Brooks DJ.: A comparison of the progression of early Parkinson’s disease in patients started on ropinirole or L-dopa: an 18F-dopa PET study. J Neural Transm. 2002 Dec; 109(12): 1433–43.
12. Parkinson Study Group. A randomized controlled trial comparing pramipexole with levodopa in early Parkinson’s disease: design and methods of the CALM-PD Study. Clin Neuropharmacol. 2000 Jan–Feb; 23(1): 34–44.
13. Checkoway H, Powers K, Smith-Weller T, Franklin GM, Longstreth WT Jr, Swanson PD.: Parkinson’s disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am J Epidemiol. 2002 Apr 15; 155(8): 732–8.
14. Tan EK, Tan C, Fook-Chong SM, Lum SY, Chai A, Chung H, Shen H, Zhao Y, Teoh ML, Yih Y, Pavanni R, Chandran VR, Wong MC.: Dose-dependent protective effect of coffee, tea, and smoking in Parkinson’s disease: a study in ethnic Chinese. J Neurol Sci. 2003 Dec 15; 216(1): 163–7.
15. Hu G, Bidel S, Jousilahti P, Antikainen R, Tuomilehto J.: Coffee and tea consumption and the risk of Parkinson’s disease. Mov Disord. 2007 Nov 15; 22(15): 2242–8.
16. Kragh CL, Lund LB, Febbraro F, Hansen HD, Gai WP, El-Agnaf O, Richter-Landsberg C, Jensen PH.: Alpha-synuclein aggregation and Ser129 phosphorylation-dependent cell death in oligodendroglial cells. Journal of Biological Chemistry; 2009; 284: 10211–22.
17. Schwarzschild MA, Schwid SR, Marek K, Watts A, Lang AE, Oakes D, Shoulson I, Ascherio A; Parkinson Study Group PRECEPT Investigators, Hyson C, Gorbold E, Rudolph A, Kieburtz K, Fahn S, Gauger L, Goetz C, Seibyl J, Forrest M, Ondrasik J.: Serumurate as a predictor of clinical and radiographic progression in Parkinson disease. Arch Neurol. 2008 Jun; 65(6): 716–23.
18. Meredith GE, Totterdell S, Potashkin JA, Surmeier DJ.: Modeling PD pathogenesis in mice: advantages of a chronic MPTP protocol. Parkinsonism Relat Disord. 2008; 14 Suppl 2: S112–5. doi: 10.1016/ j.parkreldis.2008.04.012. Epub 2008 Jun 27.
19. Choi DK, Pennathur S, Perier C, Tieu K, Teismann P, Wu DC, JacksonLewis V, Vila M, Vonsattel JP, Heinecke JW, Przedborski S.: Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J Neurosci. 2005 Jul 13; 25(28): 6594–600.
20. Etienne C Hirsch, Stéphane Hunot: Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 2009; 8: 382–97.