

Transkript
FORTBILDUNG
IgG4-assoziierte Erkrankungen
Die IgG4-assoziierte Erkrankung wurde erstmals im Jahr 2001 im Rahmen einer autoimmunen Pankreatitis beschrieben, bei der erhöhte IgG4-Serumspiegel und eine Gewebsinfiltration von IgG4-positiven Plasmazellen feststellbar waren. Im Jahr 2003 erfolgte die Definition als eigenständiges Krankheitsbild. Es handelt sich um eine inflammatorisch-fibrotische Systemerkrankung, ähnlich einer Sarkoidose, die bis heute bereits in praktisch jedem Organ beschrieben wurde.
MAJA WÜEST UND PETER HÄUSERMANN
Maja Wüest
Fallvorstellung
Eine 46-jährige Patientin stellte sich bei uns an der Dermatologischen Poliklinik erstmals im Jahr 2000 vor, weil seit 1999 juckende Papeln und Plaques im oberen Rückenbereich bestanden, die sich im Verlauf auf die Extremitäten ausbreiteten. Die Biopsie zeigte damals eine dermale Infiltration von Lymphozyten und Plasmazellen. Seit 2007 litt die Patientin an rezidivierenden, juckenden, periorbitalen Schwellungen. Im MRI des Schädels stellte sich ein Pseudotumor der Orbitae beidseits dar mit vergrösserten Tränendrüsen. Eine Biopsie der Orbita zeigte eine chronische, unspezifische, fibrosierte Dakryoadenitis ohne Hinweis für ein Lymphom, am ehesten vereinbar mit einer systemischen Autoimmunkrankheit (Kollagenose). Ein Sjögren-Syndrom konnte laborchemisch sowie histologisch nicht nachgewiesen werden. Bei vermuteter «collagen vascular disease» wurde von 2007 bis 2011 eine Systemtherapie mit Methotrexat und Prednison durchgeführt. Damit konnte ein partielles Ansprechen der Beschwerden erreicht werden. Im Jahr 2013 wurde die Patientin nochmals interdisziplinär evaluiert, da sie neu eine generalisierte Lymphadenopathie entwickelte. Es wurde erneut eine Biopsie der Haut und eines Lymphknotens entnommen. In beiden Biopsien zeigte sich ein dichtes IgG4-positives lymphoplasmazelluläres Entzündungsinfiltrat. Im Rahmen eines «second look» wurde die im Jahr 2007 entnommene Biopsie der Orbita reevaluiert und bezüglich der Verteilung der Plasmazellen untersucht, wobei sich retrospektiv ebenfalls eine IgG4-Prädominanz zeigte. Somit handelte es sich bei der periorbitalen Schwellung,
welche seit dem Jahr 2007 besteht, um eine IgG4assoziierte, autoimmune, fibrosierende Dakryoadenitis. Die Immunelektrophorese wies eine Hypergammaglobulinämie von 28,4 g/l auf (Norm: 5,6–15 g/l). In der weiteren Differenzierung der Immunglobuline war das Gesamt-IgG erhöht mit 30,5 g/l (Norm: 7–16 g/l) und ebenso das IgG4 mit > 5 g/l (Norm: 0,005–1,25 g/l). Klinisch präsentierte sich die Patientin mit persistierenden, ausgeprägten, juckenden, erythematösen Plaques und Papeln am Stamm und an den Extremitäten. Aufgrund der vorliegenden anamnestischen, klinischen, histologischen und laborchemischen Befunde konnte die definitive Diagnose einer IgG4-assoziierten Erkrankung mit Dakryoadenitis und Lymphadenopathie mit Hautbeteiligung gestellt werden.
Geschichte und Epidemiologie
Bei der IgG4-assoziierten Erkrankung handelt es sich um eine sehr seltene, chronisch entzündliche Erkrankung, die – wie bereits erwähnt – jedes Organ befallen kann. Viele initial einem Organ eigenständig zugeordnete Erkrankungen (z.B. das Mikulicz-Syndrom mit einer entzündlichen Schwellung der Tränen- und Speicheldrüsen; der Küttner-Tumor, der eine chronische lymphozytäre und sklerosierende Sialadenitis der Glandula submandibularis darstellt; die RiedelThyreoiditis, bei der es zu einer entzündlich fibrosierenden Umwandlung der Schilddrüse kommt) werden neu unter dem Begriff der IgG4-assoziierten Erkrankungen zusammengefasst. Die Ätiologie ist bis anhin nicht geklärt. Zur Inzidenz dieser Krankheit lassen sich noch keine Angaben machen, weil es sich um ein noch nicht allzu lang (seit 2003) definiertes
SZD 1/2014
23
IgG4-assoziierte Erkrankungen
schen Komplementkaskade. Das ist auch der Grund, weshalb das IgG4 eine untergeordnete Rolle in der Immunabwehr spielt. IgG4 hat die Fähigkeit, sich in der Mitte zu spalten, sodass zwei halbe Immunglobuline mit je einer Schwer- und Leichtkettenkomponente entstehen. Diese Immunglobulinhälften aggregieren wieder zusammen mit anderen Immunglobulinhälften zu neuen bispezifischen IgG4Molekülen, welche dann aber nicht mehr fähig sind, einen Crosslink zwischen Antigenen herzustellen. So geht die Fähigkeit zur Formation eines funktionierenden Immunkomplexes verloren.
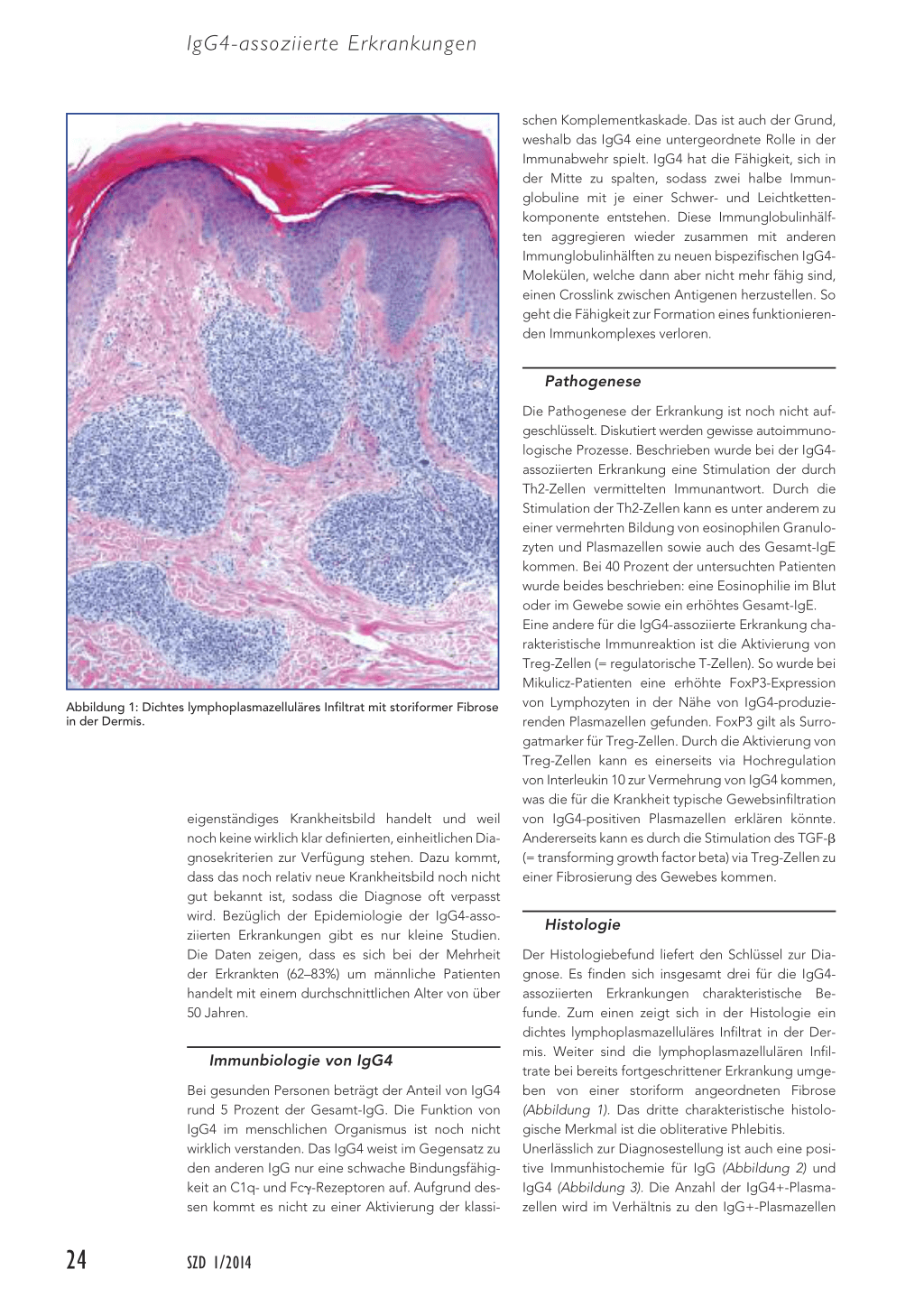
Abbildung 1: Dichtes lymphoplasmazelluläres Infiltrat mit storiformer Fibrose in der Dermis.
eigenständiges Krankheitsbild handelt und weil noch keine wirklich klar definierten, einheitlichen Diagnosekriterien zur Verfügung stehen. Dazu kommt, dass das noch relativ neue Krankheitsbild noch nicht gut bekannt ist, sodass die Diagnose oft verpasst wird. Bezüglich der Epidemiologie der IgG4-assoziierten Erkrankungen gibt es nur kleine Studien. Die Daten zeigen, dass es sich bei der Mehrheit der Erkrankten (62–83%) um männliche Patienten handelt mit einem durchschnittlichen Alter von über 50 Jahren.
Immunbiologie von IgG4
Bei gesunden Personen beträgt der Anteil von IgG4 rund 5 Prozent der Gesamt-IgG. Die Funktion von IgG4 im menschlichen Organismus ist noch nicht wirklich verstanden. Das IgG4 weist im Gegensatz zu den anderen IgG nur eine schwache Bindungsfähigkeit an C1q- und Fc␥-Rezeptoren auf. Aufgrund dessen kommt es nicht zu einer Aktivierung der klassi-
Pathogenese
Die Pathogenese der Erkrankung ist noch nicht aufgeschlüsselt. Diskutiert werden gewisse autoimmunologische Prozesse. Beschrieben wurde bei der IgG4assoziierten Erkrankung eine Stimulation der durch Th2-Zellen vermittelten Immunantwort. Durch die Stimulation der Th2-Zellen kann es unter anderem zu einer vermehrten Bildung von eosinophilen Granulozyten und Plasmazellen sowie auch des Gesamt-IgE kommen. Bei 40 Prozent der untersuchten Patienten wurde beides beschrieben: eine Eosinophilie im Blut oder im Gewebe sowie ein erhöhtes Gesamt-IgE. Eine andere für die IgG4-assoziierte Erkrankung charakteristische Immunreaktion ist die Aktivierung von Treg-Zellen (= regulatorische T-Zellen). So wurde bei Mikulicz-Patienten eine erhöhte FoxP3-Expression von Lymphozyten in der Nähe von IgG4-produzierenden Plasmazellen gefunden. FoxP3 gilt als Surrogatmarker für Treg-Zellen. Durch die Aktivierung von Treg-Zellen kann es einerseits via Hochregulation von Interleukin 10 zur Vermehrung von IgG4 kommen, was die für die Krankheit typische Gewebsinfiltration von IgG4-positiven Plasmazellen erklären könnte. Andererseits kann es durch die Stimulation des TGF- (= transforming growth factor beta) via Treg-Zellen zu einer Fibrosierung des Gewebes kommen.
Histologie
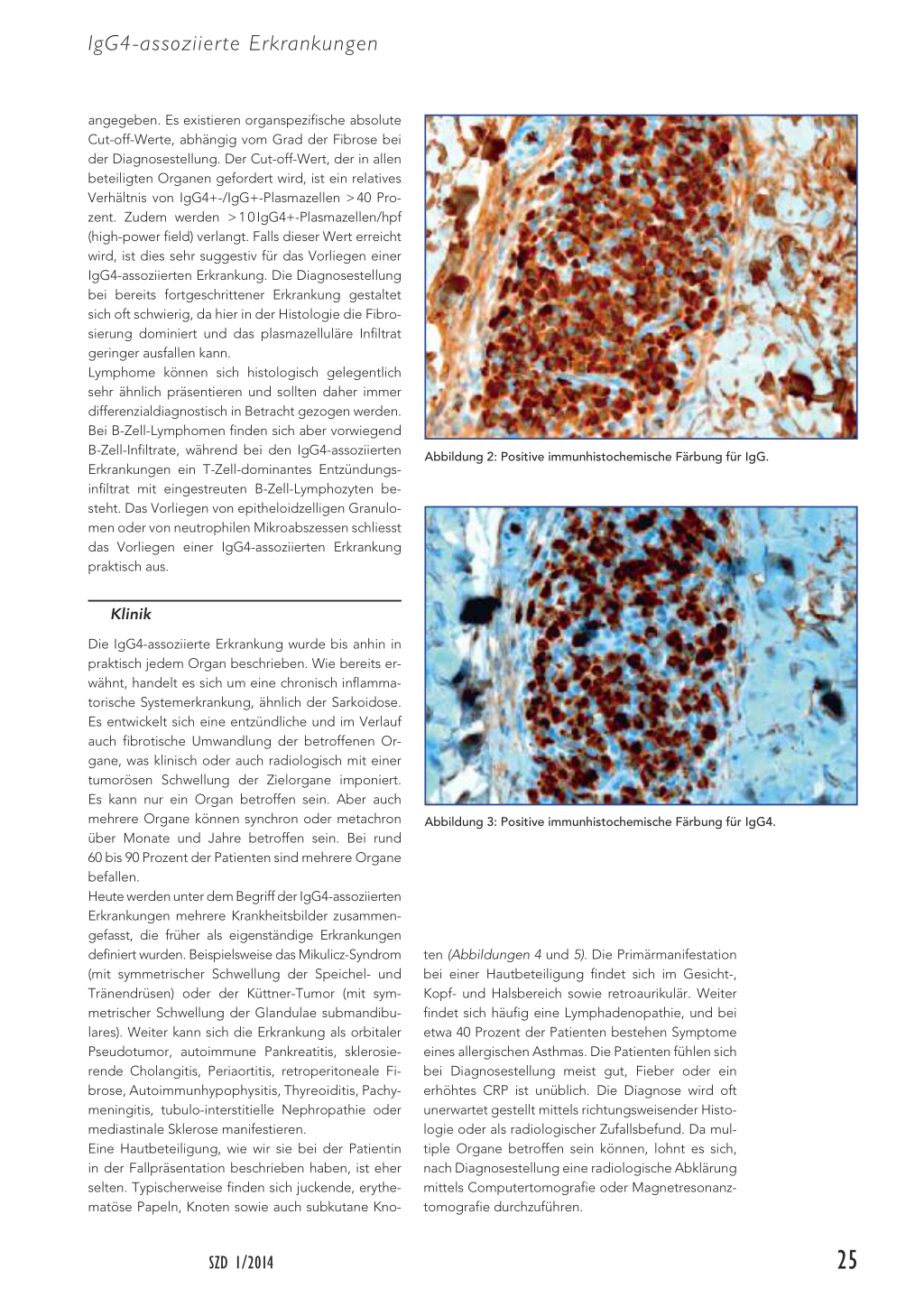
Der Histologiebefund liefert den Schlüssel zur Diagnose. Es finden sich insgesamt drei für die IgG4assoziierten Erkrankungen charakteristische Befunde. Zum einen zeigt sich in der Histologie ein dichtes lymphoplasmazelluläres Infiltrat in der Dermis. Weiter sind die lymphoplasmazellulären Infiltrate bei bereits fortgeschrittener Erkrankung umgeben von einer storiform angeordneten Fibrose (Abbildung 1). Das dritte charakteristische histologische Merkmal ist die obliterative Phlebitis. Unerlässlich zur Diagnosestellung ist auch eine positive Immunhistochemie für IgG (Abbildung 2) und IgG4 (Abbildung 3). Die Anzahl der IgG4+-Plasmazellen wird im Verhältnis zu den IgG+-Plasmazellen
24 SZD 1/2014
IgG4-assoziierte Erkrankungen
angegeben. Es existieren organspezifische absolute Cut-off-Werte, abhängig vom Grad der Fibrose bei der Diagnosestellung. Der Cut-off-Wert, der in allen beteiligten Organen gefordert wird, ist ein relatives Verhältnis von IgG4+-/IgG+-Plasmazellen > 40 Prozent. Zudem werden > 1 0 IgG4+-Plasmazellen/hpf (high-power field) verlangt. Falls dieser Wert erreicht wird, ist dies sehr suggestiv für das Vorliegen einer IgG4-assoziierten Erkrankung. Die Diagnosestellung bei bereits fortgeschrittener Erkrankung gestaltet sich oft schwierig, da hier in der Histologie die Fibrosierung dominiert und das plasmazelluläre Infiltrat geringer ausfallen kann. Lymphome können sich histologisch gelegentlich sehr ähnlich präsentieren und sollten daher immer differenzialdiagnostisch in Betracht gezogen werden. Bei B-Zell-Lymphomen finden sich aber vorwiegend B-Zell-Infiltrate, während bei den IgG4-assoziierten Erkrankungen ein T-Zell-dominantes Entzündungsinfiltrat mit eingestreuten B-Zell-Lymphozyten besteht. Das Vorliegen von epitheloidzelligen Granulomen oder von neutrophilen Mikroabszessen schliesst das Vorliegen einer IgG4-assoziierten Erkrankung praktisch aus.
Abbildung 2: Positive immunhistochemische Färbung für IgG.
Klinik
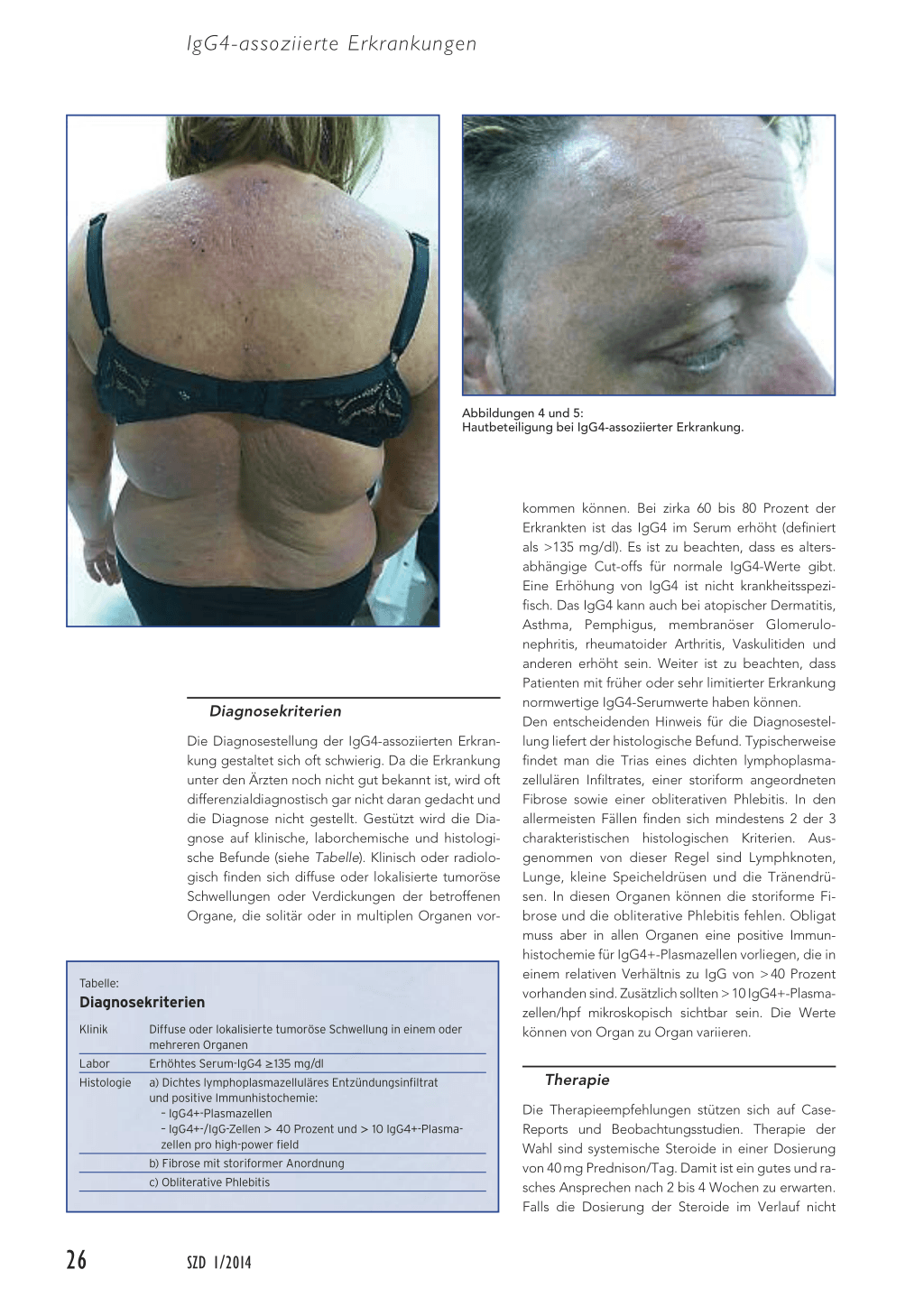
Die IgG4-assoziierte Erkrankung wurde bis anhin in praktisch jedem Organ beschrieben. Wie bereits erwähnt, handelt es sich um eine chronisch inflammatorische Systemerkrankung, ähnlich der Sarkoidose. Es entwickelt sich eine entzündliche und im Verlauf auch fibrotische Umwandlung der betroffenen Organe, was klinisch oder auch radiologisch mit einer tumorösen Schwellung der Zielorgane imponiert. Es kann nur ein Organ betroffen sein. Aber auch mehrere Organe können synchron oder metachron über Monate und Jahre betroffen sein. Bei rund 60 bis 90 Prozent der Patienten sind mehrere Organe befallen. Heute werden unter dem Begriff der IgG4-assoziierten Erkrankungen mehrere Krankheitsbilder zusammengefasst, die früher als eigenständige Erkrankungen definiert wurden. Beispielsweise das Mikulicz-Syndrom (mit symmetrischer Schwellung der Speichel- und Tränendrüsen) oder der Küttner-Tumor (mit symmetrischer Schwellung der Glandulae submandibulares). Weiter kann sich die Erkrankung als orbitaler Pseudotumor, autoimmune Pankreatitis, sklerosierende Cholangitis, Periaortitis, retroperitoneale Fibrose, Autoimmunhypophysitis, Thyreoiditis, Pachymeningitis, tubulo-interstitielle Nephropathie oder mediastinale Sklerose manifestieren. Eine Hautbeteiligung, wie wir sie bei der Patientin in der Fallpräsentation beschrieben haben, ist eher selten. Typischerweise finden sich juckende, erythematöse Papeln, Knoten sowie auch subkutane Kno-
Abbildung 3: Positive immunhistochemische Färbung für IgG4.
ten (Abbildungen 4 und 5). Die Primärmanifestation bei einer Hautbeteiligung findet sich im Gesicht-, Kopf- und Halsbereich sowie retroaurikulär. Weiter findet sich häufig eine Lymphadenopathie, und bei etwa 40 Prozent der Patienten bestehen Symptome eines allergischen Asthmas. Die Patienten fühlen sich bei Diagnosestellung meist gut, Fieber oder ein erhöhtes CRP ist unüblich. Die Diagnose wird oft unerwartet gestellt mittels richtungsweisender Histologie oder als radiologischer Zufallsbefund. Da multiple Organe betroffen sein können, lohnt es sich, nach Diagnosestellung eine radiologische Abklärung mittels Computertomografie oder Magnetresonanztomografie durchzuführen.
SZD 1/2014
25
IgG4-assoziierte Erkrankungen
Abbildungen 4 und 5: Hautbeteiligung bei IgG4-assoziierter Erkrankung.
Diagnosekriterien
Die Diagnosestellung der IgG4-assoziierten Erkrankung gestaltet sich oft schwierig. Da die Erkrankung unter den Ärzten noch nicht gut bekannt ist, wird oft differenzialdiagnostisch gar nicht daran gedacht und die Diagnose nicht gestellt. Gestützt wird die Diagnose auf klinische, laborchemische und histologische Befunde (siehe Tabelle). Klinisch oder radiologisch finden sich diffuse oder lokalisierte tumoröse Schwellungen oder Verdickungen der betroffenen Organe, die solitär oder in multiplen Organen vor-
Tabelle:
Diagnosekriterien
Klinik
Labor Histologie
Diffuse oder lokalisierte tumoröse Schwellung in einem oder mehreren Organen
Erhöhtes Serum-IgG4 ≥135 mg/dl a) Dichtes lymphoplasmazelluläres Entzündungsinfiltrat und positive Immunhistochemie:
– IgG4+-Plasmazellen – IgG4+-/IgG-Zellen > 40 Prozent und > 10 IgG4+-Plasmazellen pro high-power field
b) Fibrose mit storiformer Anordnung
c) Obliterative Phlebitis
kommen können. Bei zirka 60 bis 80 Prozent der Erkrankten ist das IgG4 im Serum erhöht (definiert als >135 mg/dl). Es ist zu beachten, dass es altersabhängige Cut-offs für normale IgG4-Werte gibt. Eine Erhöhung von IgG4 ist nicht krankheitsspezifisch. Das IgG4 kann auch bei atopischer Dermatitis, Asthma, Pemphigus, membranöser Glomerulonephritis, rheumatoider Arthritis, Vaskulitiden und anderen erhöht sein. Weiter ist zu beachten, dass Patienten mit früher oder sehr limitierter Erkrankung normwertige IgG4-Serumwerte haben können. Den entscheidenden Hinweis für die Diagnosestellung liefert der histologische Befund. Typischerweise findet man die Trias eines dichten lymphoplasmazellulären Infiltrates, einer storiform angeordneten Fibrose sowie einer obliterativen Phlebitis. In den allermeisten Fällen finden sich mindestens 2 der 3 charakteristischen histologischen Kriterien. Ausgenommen von dieser Regel sind Lymphknoten, Lunge, kleine Speicheldrüsen und die Tränendrüsen. In diesen Organen können die storiforme Fibrose und die obliterative Phlebitis fehlen. Obligat muss aber in allen Organen eine positive Immunhistochemie für IgG4+-Plasmazellen vorliegen, die in einem relativen Verhältnis zu IgG von > 40 Prozent vorhanden sind. Zusätzlich sollten > 10 IgG4+-Plasmazellen/hpf mikroskopisch sichtbar sein. Die Werte können von Organ zu Organ variieren.
Therapie
Die Therapieempfehlungen stützen sich auf CaseReports und Beobachtungsstudien. Therapie der Wahl sind systemische Steroide in einer Dosierung von 40 mg Prednison/Tag. Damit ist ein gutes und rasches Ansprechen nach 2 bis 4 Wochen zu erwarten. Falls die Dosierung der Steroide im Verlauf nicht
26 SZD 1/2014
IgG4-assoziierte Erkrankungen
unter die Cushing-Schwellendosis reduziert werden kann, müssen als Zweitlinientherapie steroidsparende Substanzen wie Azathioprin oder Mycophenolatmofetil eingesetzt werden. Weitere Fallberichte von Arezou Khosroshai et al. beschreiben auch ein gutes Ansprechen auf Rituximab. Auch unsere Patientin musste im Verlauf auf Rituximab umgestellt werden. Die Patientin hatte zwar ein gutes Ansprechen auf Prednison, das aber nicht unter die CushingSchwelle reduziert werden konnte bei erneuten Rezidiven der Grunderkrankung. Zudem entwickelte die Patientin einen steroidinduzierten Diabetes. Unter einer Therapie mit Rituximab zeigte die Patientin dann im Verlauf eine Remission der Beschwerden. Es gibt Beschreibungen von Spontanremissionen bei der IgG4-assoziierten Erkrankung. Es sind aber auch Rezidive nach spontanen Remissionen und nach erfolgreicher Steroidtherapie beschrieben worden. Bei lang bestehender, unbehandelter Erkrankung kann es zu einer starken fibrotischen Umwandlung der betroffenen Organe kommen, was gegebenenfalls eine Organdysfunktion zur Folge haben kann.
Zusammenfassung
Die IgG4-assoziierte Erkrankung ist eine chronisch entzündliche Systemerkrankung, die eines oder mehrere Organe befallen kann, ähnlich einer Sarkoidose. Klinisch imponiert eine entzündliche tumoröse Schwellung der betroffenen Organe. Eine Hautbetei-
ligung ist selten. Den Schlüssel zur Diagnose liefert die charakteristische Histologie mit der Trias eines lymphoplasmazellulären Entzündungsinfiltrates, einer storiform angeordneten Fibrose sowie dem Vorliegen einer obliterativen Phlebitis. Unerlässlich für die Diagnose ist auch eine positive Immunhistochemie für IgG4-Plasmazellen. Generell besteht ein gutes Ansprechen auf Immunsuppressiva. Die Diagnose wird oft verkannt, da es sich um ein noch nicht allzu lang definiertes Krankheitsbild handelt. Oft wird deshalb differenzialdiagnostisch nicht daran gedacht. L
Kontaktadresse: Dr. med. Maja Wüest Dermatologie Universitätsspital Basel Petersgraben 4 4031 Basel E-Mail: Maja.Wueest@usb.ch
Interessenkonflikte: keine
Literaturangaben: Stone JH et al. IgG4-Related Disease. N Engl J Med 2012; 366: 539–551. Deshpande V et al. Consensus statement on the pathology of IgG4-related disease. Modern Pathology 2012; 25: 1181–1192. Ingen-Housz-Oro S et al. IgG4-related skin disease successfully treated with thalidomide. JAMA Dermatol 2013; 149: 742–747. Yamamoto M et al. Value of serum IgG4 in the diagnosis of IgG4related disease and in differentiation for rheumatic disease and other diseases. Mod Rheumatol 2012; 22: 419–425. Umehara H et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol 2012; 22: 1–14.
SZD 1/2014
27