



Transkript
HAUTKREBS
Pathogenese epithelialer Hauttumoren
Vortrag von PD Dr. med. Werner Kempf anlässlich der Fortbildungsveranstaltung der Universität Zürich «Epitheliale Hauttumoren – Neues zur Prävention und Therapie» am 31. Januar 2008.
Neuere wissenschaftliche Erkenntnisse ermöglichen dank Fortschritten in Immunologie und Molekulargenetik vermehrt Einblick in Ursachen und Entstehung von Hauttumoren. Histopathologische Untersuchungen werden zusätzlich durch Erkenntnisse aus dem genetischen Profil der Tumoren ergänzt, was neben Aussagen zur Diagnose auch solche zur Prognose und zu therapeutischen Vorgehensweisen erlauben wird.
rung der Mitose- und Proliferationsrate, zum anderen wird diese ebenfalls gesteigert, wenn Tumorsuppressorgene inaktiviert werden oder Mutationen vorliegen (Abbildung 1). Ein wichtiger Keyplayer ist das Tumorsuppressorgen p53, das über verschiedene Mechanismen direkt eingreift. Es ist dafür besorgt, dass DNA-Schäden repariert werden können. Ist ein Schaden irreparabel, wird die Zelle gezwungen, in den programmierten Zelltod (Apoptose) zu gehen.
Mitogene Faktoren ras MYC FOS/JUN EGFR2 BCl-2
Protoonkogene
Mitose Proliferation
Proliferationsinhibitoren p53 RB PTEN APC
Tumorsuppressorgene
Aktivierung
Tumorwachstum
Inaktivierung Mutationen
Ein Tumor kann als eine Dysbalance zwischen Proliferation von Zellen und dem programmierten Zelltod beschrieben werden. Im Normalfall sollte ein Gleichgewicht zwischen mitosefördernden und proliferationshemmenden Faktoren gewahrt sein. In diesen Prozessen sind Moleküle involviert, die entweder als Protoonkogene oder als Tumorsuppressorgene bezeichnet werden. Bei der Tumorentstehung kommt es zu einem Ungleichgewicht zwischen diesen Mitspielern. Zum einen führt die Aktivierung von Protoonkogenen zu einer Steige-
Abbildung 1: Neoplasie – Dysbalance von Proliferation und Apoptose
Die Tumorbildung wird nicht durch einen einzigen Faktor ausgelöst, sondern es sind mehrere genetische Veränderungen notwendig, die in einer bestimmten Sequenz in den einzelnen Tumoren ablaufen. Daher wird nach Vogelstein und Kinzler (Nat Med 2004) die Entstehung eines Tumors als ein evolutionärer Prozess verstanden, der durch genetische Mutationen vorangetrieben wird.
21
medicos 2/2008
HAUTKREBS
UV-Strahlung und Hauttumore
UV-B-Licht führt zu Tumorsuppressorgen-Mutationen in den Keratinozyten. Es kommt zum Funktionsverlust von p53, was die Tür zur unkontrollierten Proliferation der Keratinozyten und Apoptoseinhibition öffnet (Abbildung 2). In Hauttumoren epithelialer Genese finden wir zu einem sehr hohen Prozentsatz UV-B-induzierte, inaktivierende Mutationen im p53-Gen: 53 bis 66 Prozent in aktinischen Keratosen und 35 bis 58 Prozent in spinozellären Karzinomen. Somit ist die UV-B-Strahlung ein ganz zentraler Faktor für die Tumorentstehung.
UV-B-Strahlung
p53-Mutationen in Keratinozyten
p-53-Funktionverlust
Induktion und Hochregulation des Epidermal Growth Factor Receptor EGFR
dann normalerweise ab dem 50. bis 60. Lebensjahr zu manifesten Tumoren kommen kann. Bei Xeroderma-pigmentosum-Patienten kommt der zweite «hit» schon sehr früh im Leben, meist in der Adoleszenz. Kommen im Verlauf des Lebens weitere Mutationen wie zum Beispiel in p53-, NER- oder rasGenen hinzu, entstehen sehr früh im Leben erschreckende Bilder: spinozelluläre Karzinome und Tumoren der viszeralen Organe. 70 Prozent dieser Patienten überleben das 40. Lebensjahr nicht.
Bedeutung von Wachstumsfaktoren Es kommen Regulationsmechanismen dazu, die nicht auf genetischer Ebene determiniert sind. Damit ein Tumor wachsen kann, werden Wachstumsfaktoren benötigt. Ein Beispiel ist der Epidermal Growth Factor, dessen Wirkung über einen Rezeptor mediiert wird (Abbildung 2). Dieser Rezeptor lässt sich blockieren, beispielsweise durch einen Antikörper, der zur kompetitiven Hemmung des Rezeptorsignals führt. Aus diesem biologischen Verständnis heraus sind einige Therapeutika, wie zum Beispiel Cetuximab (Erbitux®), entwickelt worden.
Proliferation Apoptoseinhibition
Immunsuppression und Hauttumore
Nicht nur Patienten mit Xeroderma pigmentosum,
Abbildung 2: Einfluss der UV-B-Strahlung
sondern auch Transplantatempfänger zeigen eine deutlich erhöhte Inzidenz von epithelialen Haut-
tumoren. Diese nimmt mit der Dauer der Immun-
Genodermatosen als Modellerkrankungen suppression zu, sodass zehn Jahre nach der Trans-
Bei genetischen Syndromen wie zum Beispiel beim plantation bereits bis zu 70 Prozent der Patienten an
Xeroderma pigmentosum liegt eine besondere Situa- Hautkrebs leiden. Hier steht bezüglich der Patho-
tion vor. Sie dienen deshalb als Modellerkrankung genese nicht primär die Genetik im Vordergrund,
für das bessere Verständnis der Pathogenese von sondern die Immunsuppression, das heisst offen-
Hauttumoren. Xeroderma pigmentosum ist durch sichtlich die eingeschränkte Funktion eines intakten
eine erhöhte Fotosensitivität, einen Reparaturdefekt Immunsystems.
bei UV-induzierten Schäden und, aus diesen Fakto- Zusätzlich spielen auch medikamentöse Einflüsse
ren resultierend, durch ein hohes Hautkrebsrisiko eine wichtige Rolle. Sie können die UV-A-bedingte
gekennzeichnet. Davon betroffene Patienten sind DNA-Schädigung verstärken und reaktive Sauerstoff-
sehr viel früher mit Tumoren konfrontiert als spezies induzieren. Azathioprin zum Beispiel erhöht
andere, da Mutationen bereits direkt in der Keim- die Empfindlichkeit gegenüber UV-A-Strahlung, wel-
bahn vorliegen. Der gesamte komplexe DNA-Repa- che direkt mutagene Eigenschaften aufweist.
raturmechanismus ist gestört: Die geschädigte DNA Bekannt ist dieses Phänomen ausserhalb der Immun-
sollte aufgerollt werden, damit die Reparatur- suppression beim sogenannten Keratoakanthom. Es
enzyme so arbeiten, dass sie die defekten Teile handelt sich hierbei um rasch aufschiessende Tumo-
herausschneiden können, welche danach durch ren mit zentralen Kratern. Man geht davon aus, dass
medicos 2/2008
DNA-Polymerase wieder ersetzt werden.
die Rückbildung dieser Tumoren entscheidend von
In den frühen Siebzigerjahren wurde die Hypothese der Immunantwort abhängt. Die Tumorzellen indu-
aufgestellt, dass eine genetische Mutation allein zieren eine Immunantwort, die ganz wesentlich über
nicht ausreicht, sondern dass es mehrere Ereignisse dendritische Zellen mediiert wird, aber auch über
(hits) zur Tumorbildung braucht. Heute wissen wir, T-Lymphozyten. Diese Zellen versuchen ihrerseits,
dass in der Regel mindestens fünf benötigt werden. den Tumor zu eliminieren. Hier liegt ein interes-
Das bedeutet, dass über die Zeit unseres Lebens santer Ansatz für immunaktivierende Therapien zur
22 genetische Mutationen akkumuliert werden, und es Tumorbehandlung. Die Erfahrung zeigt, dass die
HAUTKREBS
antitumorale Immunantwort meistens ineffizient ist, wenn sie nicht verstärkt wird. Hier ist ein Angriffspunkt für Agonisten der Toll-like-Rezeptoren wie Imiquimod oder auch Katechine (Polyphenone, Grünteeextrakt). Zum Verständnis der Wirkung von Imiquimod haben Arbeitsgruppen von Frau PD Dr. M. Urosevic und Prof. Dr. R. Dummer Wesentliches beigetragen: Unter Therapie ist nach drei bis fünf Tagen ein Infiltrat aus Makrophagen und Lymphozyten und eine verringerte BCl-2-Expression der Tumorzellen zu beobachten. Unter Imiquimod werden nicht nur die Entzündungszellen reduziert, sondern es wird direkt die Apoptose induziert, was mit der verminderten Expression von anti-apoptotischen Molekülen wie BCl-2 korreliert.
Entzündung als tumorpropagierender Faktor Was in der akuten Form ein Benefit sein kann, kann bei einem chronischen Verlauf genau das Gegenteil bewirken. Ein Hinweis, dass chronische Entzündungen ein tumorpropagierender Faktor sein können, ist beispielsweise die erhöhte Inzidenz von spinozellulären Karzinomen bei Lichen ruber. Ebenso gibt es Studien, die belegen, dass die aktinischen Keratosen, unter denen sich eine Entzündung befindet, ein erhöhtes Risiko haben, in ein invasives spinozelluläres Karzinom überzugehen. Eine Ursache dieses Phänomens ist das unterschiedliche Zytokinmuster von akuten und chronischen Entzündungen (Abbildung 3).
akute Entzündung Th1 B-Zelle
chronische Entzündung Treg Th2 B-Zelle
IL-2 Antitumor Ig INF␥
IL-10 TGF
IL-4 IL-6 IL-10 IL-10 IL-13 Stroma Ig/IC
M1 Makrophagen Polarität Antitumor Zellzytoxizität CTL-induzierte Zerstörung
Tumorabstossung
M2 Makrophagen Polarität Antigenpräsentation CTL-induzierte Zerstörung
Tumorpromotion
Abbildung 3: Unterschiedliches Zytokinmuster bei akuter und chronischer Entzündung (aus: DeNardo & Coussens, Breast Cancer Res. 2007; 9: 212.)
Th2-Zytokine haben bei chronischen Entzündungen einen tumorpromovierenden Effekt. Aus therapeutischer Sicht ist dies ein Angriffspunkt für die Entzündungshemmung mittels nichtsteroidaler, antiinflammatorischer Medikamente wie zum Beispiel Diclofenac (Solaraze-Gel®) bei aktinischen Keratosen.
Humane Papillomviren und Tumore
Der dritte Faktor in der Tumorpathogenese epithelialer Hauttomoren sind Viren, welche direkt Tumore induzieren können (Abbildung 4). Wichtig sind die Hochrisikotypen der humanen Papillomviren (HPV 16, 18, 31, 33, 35, usw.). Diese tragen selber zwei Onkogene (E6, E7), die ihrerseits über zelluläre Onkogene zur Tumorentstehung beitragen. Sie spielen eine wichtige Rolle bei den anogenitalen Karzinomen und deren Vorstufen, aber auch bei gewissen Subtypen enoraler Plattenepithelkarzinome. In Zukunft werden Vakzine gegen diese Hochrisikovirentypen einen wichtigen Beitrag leisten können. Aber 10 Prozent der Tumoren werden durch andere HPV-Viren induziert, für welche noch keine Impfstoffe zur Verfügung stehen.
Chemische Karzinogene
Vorläuferzelle
UV-Licht
Genetische Alterationen Transformation
Onkogene Viren
Tumor
Abbildung 4: Pathogenesefaktoren epithelialer Hauttumoren
Interessanterweise sind für die Haut nicht die erwähnten anogenitalen HPV-Typen relevant, sondern kutane Typen, die sogenannten Beta-HPVTypen, die bei der Epidermodysplasia verruciformis auftreten. Bei dieser Erkrankung bilden sich mit einem genetischen Hintergrund epidermale Neoplasien aufgrund von Infektionen durch die kutanen HPV-Typen. Sie kommen sehr häufig auch bei immunkompetenten Patienten in epithelialen Tumoren wie aktinischen Keratosen vor. Können kutane HPV-Typen nachgewiesen werden, haben entsprechende Patienten ein erhöhtes Risiko, an aktinischen Keratosen zu erkranken.
Diagnostische Marker
Für die diagnostische Praxis ist die Kenntnis genetischer Alterationen sehr hilfreich. So kann zum Beispiel aufgrund der Expression des Tumorsuppressorgens p16 ein Morbus Bowen von einer seborrhoischen Keratose abgegrenzt werden: p16Expression tritt zu 84 Prozent in M. Bowen, aber nur zu 7 Prozent bei aktinischer Keratose auf. Ein Signaltumor ist das Talgdrüsenadenom, das per se zwar gutartig ist, aber sehr häufig mit dem Muir-Torre-Syndrom assoziiert ist, welches mit
medicos 2/2008
23
HAUTKREBS
gastrointestinalen und urogenitalen Karzinomen einhergeht. Es ist zentral für betroffene Patienten und ihre Familien, solche Talgdrüsenadenome als Markerläsionen rechtzeitig zu erkennen. In diesen Tumoren lässt sich heute sehr elegant immunhistochemisch der Verlust der Proteinexpression nachweisen. Ohne zusätzliche Biopsie können am vorliegenden formalinfixierten Material die Untersuchungen durchgeführt werden. Eine PCR-Untersuchung belegt die genetische Mutation in den DNA-Mismatch-Reparaturgenen und somit die Diagnose eines Muir-Torre-Syndroms. Gerade bei Patienten, die über 40 sind, sollten solche Tumore als Warnsignal betrachtet werden (Abbildung 5).
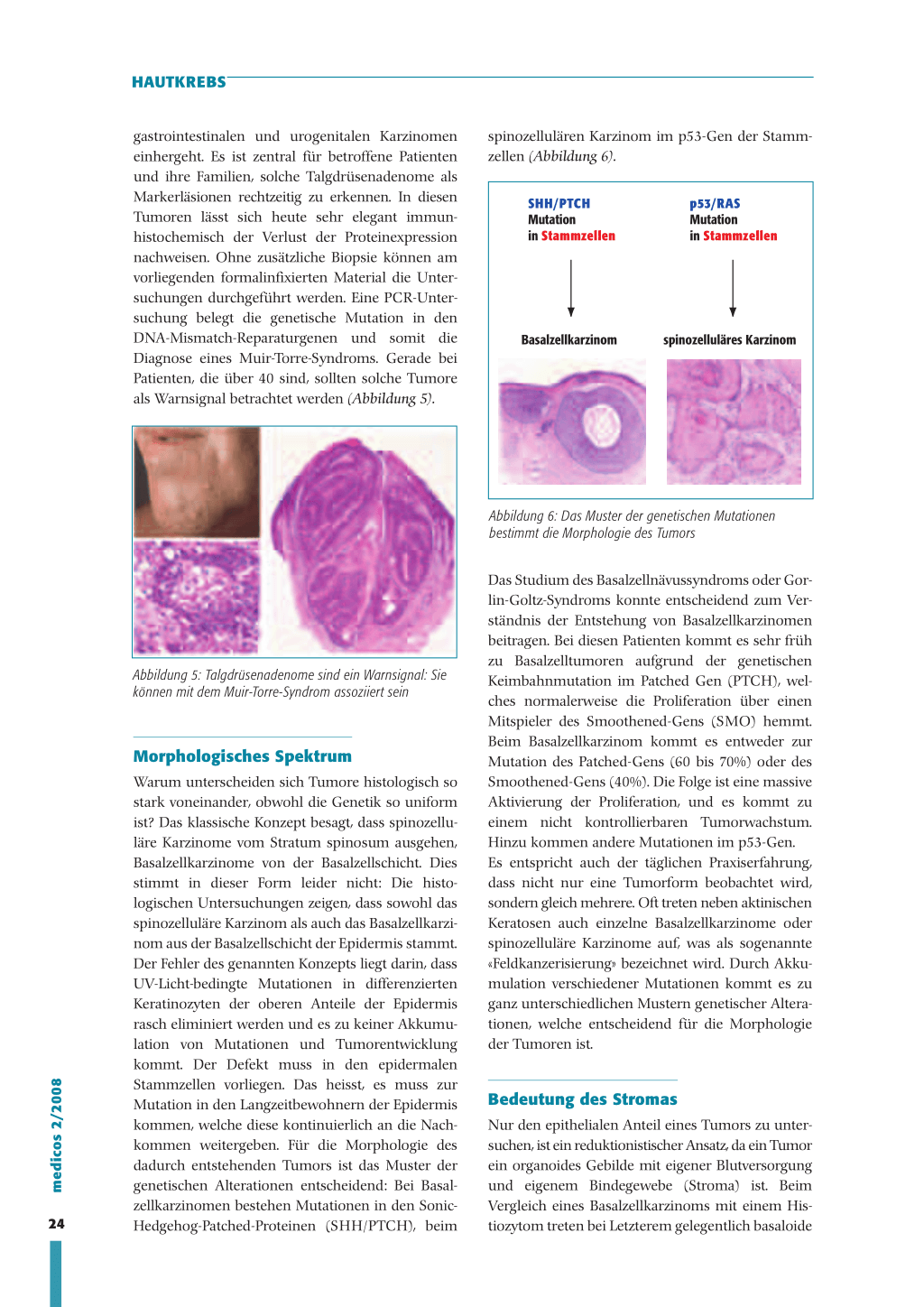
spinozellulären Karzinom im p53-Gen der Stammzellen (Abbildung 6).
SHH/PTCH Mutation in Stammzellen
p53/RAS Mutation in Stammzellen
Basalzellkarzinom
spinozelluläres Karzinom
Abbildung 6: Das Muster der genetischen Mutationen bestimmt die Morphologie des Tumors
Das Studium des Basalzellnävussyndroms oder Gor-
lin-Goltz-Syndroms konnte entscheidend zum Ver-
ständnis der Entstehung von Basalzellkarzinomen
beitragen. Bei diesen Patienten kommt es sehr früh
Abbildung 5: Talgdrüsenadenome sind ein Warnsignal: Sie können mit dem Muir-Torre-Syndrom assoziiert sein
zu Basalzelltumoren aufgrund der genetischen Keimbahnmutation im Patched Gen (PTCH), welches normalerweise die Proliferation über einen
Mitspieler des Smoothened-Gens (SMO) hemmt.
Morphologisches Spektrum
Beim Basalzellkarzinom kommt es entweder zur Mutation des Patched-Gens (60 bis 70%) oder des
Warum unterscheiden sich Tumore histologisch so Smoothened-Gens (40%). Die Folge ist eine massive
stark voneinander, obwohl die Genetik so uniform Aktivierung der Proliferation, und es kommt zu
ist? Das klassische Konzept besagt, dass spinozellu- einem nicht kontrollierbaren Tumorwachstum.
läre Karzinome vom Stratum spinosum ausgehen, Hinzu kommen andere Mutationen im p53-Gen.
Basalzellkarzinome von der Basalzellschicht. Dies Es entspricht auch der täglichen Praxiserfahrung,
stimmt in dieser Form leider nicht: Die histo- dass nicht nur eine Tumorform beobachtet wird,
logischen Untersuchungen zeigen, dass sowohl das sondern gleich mehrere. Oft treten neben aktinischen
spinozelluläre Karzinom als auch das Basalzellkarzi- Keratosen auch einzelne Basalzellkarzinome oder
nom aus der Basalzellschicht der Epidermis stammt. spinozelluläre Karzinome auf, was als sogenannte
Der Fehler des genannten Konzepts liegt darin, dass «Feldkanzerisierung» bezeichnet wird. Durch Akku-
UV-Licht-bedingte Mutationen in differenzierten mulation verschiedener Mutationen kommt es zu
Keratinozyten der oberen Anteile der Epidermis ganz unterschiedlichen Mustern genetischer Altera-
rasch eliminiert werden und es zu keiner Akkumu- tionen, welche entscheidend für die Morphologie
lation von Mutationen und Tumorentwicklung der Tumoren ist.
kommt. Der Defekt muss in den epidermalen
medicos 2/2008
Stammzellen vorliegen. Das heisst, es muss zur Mutation in den Langzeitbewohnern der Epidermis
Bedeutung des Stromas
kommen, welche diese kontinuierlich an die Nach- Nur den epithelialen Anteil eines Tumors zu unter-
kommen weitergeben. Für die Morphologie des suchen, ist ein reduktionistischer Ansatz, da ein Tumor
dadurch entstehenden Tumors ist das Muster der ein organoides Gebilde mit eigener Blutversorgung
genetischen Alterationen entscheidend: Bei Basal- und eigenem Bindegewebe (Stroma) ist. Beim
zellkarzinomen bestehen Mutationen in den Sonic- Vergleich eines Basalzellkarzinoms mit einem His-
24 Hedgehog-Patched-Proteinen (SHH/PTCH), beim tiozytom treten bei Letzterem gelegentlich basaloide
HAUTKREBS
Epithelproliferate auf, die nicht von einem superfiziellen Basalzellkarzinom zu unterscheiden sind. Beide Stromaformen, die Bindegewebsanteile dieser Tumoren also, sezernieren den Epidermal Growth Factor (EGF) und können so basaloide Proliferate induzieren. Hier liegt einer der interessanten Ansatzpunkte, welche die Diagnose im Alltag künftig revidieren könnten. Bei der Beurteilung, ob ein Basalzellkarzinom im Gesunden entfernt wurde, wird man sich nicht nur mehr auf die epithelialen Anteile konzentrieren, sondern in Zukunft vermehrt auch auf das Stroma.
Zusammenfassung
Die Tumorentstehung ist eine Kaskade genetischer Veränderungen, die eine Dysbalance zwischen Onkogenen und Tumorsuppressorgenen umfassen. Das Studium von Genodermatosen hat ganz entscheidend zum Verständnis der Pathogenese von Hauttumoren beigetragen. Das Auftreten des Talgdrüsenadenoms muss als Warnsignal betrachtet werden. Es kann mit dem Muir-Torre-Syndrom assoziiert sein. Aus den pathogenetischen Erkenntnissen lassen sich neue Therapieansätze gewinnen.
Die Morphologie von epithelialen Hauttumoren
wird klinisch und histologisch stark durch das
Mutationsmuster geprägt, eine Erkenntnis, die
aus dermatopathologischer Sicht eine vielverspre-
chende Entwicklung ermöglicht: In etwa zehn Jah-
ren sollte es technisch möglich sein, dass routine-
mässig histochemische Untersuchungen zusätzlich
durch Erkenntnisse aus dem Proteinprofil und
genetischen Profil der Tumoren ergänzt werden.
Dies wird die Arbeit des Dermatopathologen verän-
dern, da nicht mehr nur Aussagen zur Diagnose
gemacht werden können, sondern auch zur Pro-
gnose und möglicherweise zum therapeutischen
Ansprechen von Tumoren.
●
Gisela Stauber-Reichmuth
Korrespondenzadresse für inhaltliche Anfragen: PD Dr. med. Werner Kempf Kempf und Pfaltz Histologische Diagnostik, Seminarstrasse 1, 8042 Zürich Tel. 043-443 11 77, E-Mail: kempf@kempf-pfaltz.ch Konziliararzt und wissenschaftlicher Mitarbeiter Dermatologische Klinik, UniversitätsSpital Zürich
Interessenkonflikte: keine