



Transkript
Im Fokus: Neue Wege in der Krebsdiagnostik und personalisierten Medizin
Hochdurchsatz-Sequenzierung in der molekularen Tumordiagnostik
Methodik und Anwendung
Die Hochdurchsatz-Sequenzierung (Next Generation Sequencing, NGS) ermöglicht die parallele Sequenzierung von mehreren DNA-Abschnitten in einem Durchgang. Die Methode wurde in den letzten Jahren erfolgreich zur umfassenden Charakterisierung von Tumorgenomen eingesetzt. Dieser Beitrag beschreibt Anwendungen der NGS-Technologie in der klinischen Tumordiagnostik. Hier hat sich die zielgerichtete Hochdurchsatz-Sequenzierung als nützliches Werkzeug zur Untersuchung von Biomarkern für die personalisierte Krebstherapie etabliert.
SABINE BAUMANN, WOLFRAM JOCHUM
SZO 2018; 4: 6–10.
Sabine Baumann Wolfram Jochum
Der Erwerb von somatischen Mutationen ist ein Schlüsselmechanismus bei der Entstehung von soliden Tumoren und hämato-onkologischen Erkrankungen (1). Im Rahmen des Projekts «The Cancer Genome Atlas» (TCGA) wurden in den letzten Jahren 33 Tumorentitäten umfassend auf DNA- und RNAEbene charakterisiert. Die Ergebnisse wurden kürzlich als «Pan-Cancer-Atlas» veröffentlicht (2). Obwohl technologisch Sequenzierungen des gesamten Exoms oder Genoms eines Tumors und eine Expressionsanalyse des gesamten Transkriptoms durchgeführt werden können, steht in der klinischen Tumordiagnostik die zielgerichtete Sequenzierung von tumorassoziierten Genen und Fusionstranskripten im Vordergrund (3).
Tumorbiologische Grundlagen
Erworbene somatische Mutationen sind die Treiber der Tumorentstehung. Neben einfachen Sequenzvarianten (Einzelnukleotidvarianten [SNV] wie Missense/Nonsense-Mutationen, kleine Insertionen/Deletionen), die nur ein einzelnes Nukleotid oder wenige Nukleotide betreffen, werden strukturelle
ABSTRACT
High-throughput sequencing and molecular tumor diagnostics
High-throughput sequencing (next generation sequencing, NGS) has become a key technology in cancer diagnostics. Targeted sequencing approaches using panels of cancer-associated genes are used to identify mutations that can be applied as diagnostic and/or prognostic biomarkers. Other NGS applications include patient selection for targeted therapies, comprehensive mutational profiling to stratify patients for clinical trails and experimental therapies, and tumor mutational load testing. This article reviews key aspects related to NGS applications in personalized cancer medicine.
Keywords: Next generation sequencing – targeted sequencing – gene panel – mutations
und numerische Chromosomenveränderungen (Translokationen, Amplifikationen, Deletionen) unterschieden. Somatische Mutationen betreffen mehrheitlich Gene, die für Proteine kodieren, welche an intrazellulären Vorgängen wie Signalübermittlung, Zellteilung und programmiertem Zelltod (Apoptose) sowie an zellulären Prozessen wie invasivem Wachstum, Angiogenese und Tumorzell-Stroma-Interaktion beteiligt sind (1). Funktionell führen Mutationen in tumorassoziierten Genen zu einer Aktivierung oder Inaktivierung der kodierten Proteine.
Methodische Aspekte
Für die Hochdurchsatz-Sequenzierung stehen verschiedene Methoden (Sequenzierung mit Brückensynthese, Halbleitersequenzierung etc.) und Geräteplattformen zur Verfügung (4). Die wesentlichen Arbeitsschritte der klinischen Tumordiagnostik sind jedoch gleich, unabhängig davon, welches Verfahren der zielgerichteten Hochdurchsatz-Sequenzierung eingesetzt wird (Abbildung 1). Die Sequenzierung erfolgt auf DNA-Ebene. RNA-Moleküle können nach einem Umschreiben in komplementäre DNA sequenziert und quantifiziert werden (RNA-Seq). In der klinischen Tumordiagnostik werden vor allem Gengruppen (sogenannte Genpanels) von bis zu mehreren Hundert Zielregionen in tumorassoziierten Genen analysiert. Genpanels für unterschiedliche Anwendungen sind kommerziell erhältlich oder können durch den Anwender zusammengestellt werden. Entsprechend unterscheiden sich die in der klinischen Routinediagnostik verwendeten Genpanels hinsichtlich Grösse und Genzusammensetzung. Untersuchungen mit Genpanels haben im Vergleich zur Sequenzierung des gesamten Exoms/Genoms (WES/WGS) Vorteile: kürzere Sequenzierzeiten,
6 SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 4/2018
Im Fokus: Neue Wege in der Krebsdiagnostik und personalisierten Medizin
höhere Sequenziertiefe der einzelnen Regionen, schnellere Datenanalyse/-interpretation sowie einfachere Standardisierung und Qualitätssicherung. Bei der bioinformatischen Primäranalyse werden die Rohdaten des Sequenziergeräts in Sequenzdaten mit assoziierten Qualitätsmerkmalen konvertiert. Die Sekundäranalyse identifiziert Sequenzvarianten durch den Vergleich der Sequenzdaten mit den entsprechenden Abschnitten eines humanen Referenzgenoms (hg19, GRCh38). Für die Beschreibung von Sequenzvarianten steht eine standardisierte Nomenklatur der HGVS zur Verfügung (5). Durch geeignete Bioinformatikinstrumente können auch numerische und strukturelle Veränderungen von Genregionen (v.a. Amplifikationen und Translokationen) nachgewiesen werden. Alternativ können Translokationen durch die Sequenzierung der entsprechenden Fusionstranskripte detektiert werden. Die Annotation einer genomischen Veränderung beurteilt ihre funktionellen Auswirkungen und die Relevanz für die Tumorentstehung. Die Pathogenität von somatischen Sequenzvarianten wird in der Regel analog zu Keimbahnveränderungen in einer von fünf Kategorien klassifiziert (6). Informationen zur Pathogenität finden sich in allgemein zugänglichen Datenbanken (COSMIC, dSNP, 100 Genomes, ClinVar, etc.). Klinisch relevant sind vor allem pathogene Varianten, die als Treibermutationen der Tumorentstehung wirken. Diagnostisch unbedeutend sind neutrale Sequenzvarianten als Zeichen der genetischen Variabilität innerhalb und zwischen Populationen (Polymorphismen) sowie Sequenzvarianten ohne direkten Bezug zur Tumorentstehung (Passenger Mutationen). Varianten mit unbekannter Signifikanz (VUS) lassen sich aufgrund des aktuellen Wissensstands nicht als sicher pathogen oder neutral kategorisieren. Zu Beurteilung der funktionellen Konsequenzen einer Sequenzvariante werden meistens bioinformatische Vorhersagemodelle verwendet. Nur bei wenigen Sequenzvarianten beruht die Kategorisierung auf experimenteller Evidenz. Im klinischen Kontext ist vor allem die Bedeutung einer genomischen Alteration für die Patientenbetreuung zu beurteilen («actionability»). Eine genomische Alteration kann als «actionable» betrachtet werden, falls sie ein Ansprechen oder eine Resistenz auf eine zugelassene oder experimentelle Therapie voraussagt beziehungsweise als diagnostischer und/oder prognostischer Marker verwendet werden kann (7). Informationen zur Actionability einer Mutation finden sich in öffentlich zugänglichen Onlinedatenbanken wie MyCancerGenome.org, Oncokb.org oder PersonalizedCancerTherapy.org (8). Kürzlich veröffentlichte AMP/ASCO/CAP-Kriterien ermöglichen zusätzlich eine Gewichtung der verfügbaren Evidenz, um die Zuverlässigkeit der Beurteilung vor allem in Hinblick auf Therapieempfehlungen zu bewerten (8).
Abbildung: Tumormutationsprofilierung mittels zielgerichteter Hochdurchsatz-Sequenzierung. Das Flussdiagramm zeigt die wesentlichen Arbeitsschritte von der Auswahl des Probematerials bis zur Berichterstellung.
Klinische Anwendungen
Veränderungen in tumorassoziierten Genen werden als diagnostische und prognostische Biomarker sowie zur Vorhersage eines Therapieansprechens eingesetzt (Tabelle 1). Die zielgerichtete Hochdurchsatz-Sequenzierung kann an paraffineingebetteten Gewebeproben, zytologischem Probenmaterial, Knochenmarkaspiraten und Tumorzellen in Blutproben (v.a. bei Leukämien/Lymphomen) durchgeführt werden. Vor der DNA/RNA-Gewinnung ist ein ausreichender Tumorzellgehalt des Probenmaterials sicherzustellen oder eine gezielte Tumorzellanreicherung durchzuführen (z.B. durch Mikrodissektion).
Tumorklassifikation und Risikostratifizierung Aktuelle Klassifikationssysteme verwenden vor allem phänotypische Merkmale zur Tumoreinteilung. Bei der Definition und Namensgebung von Tumorentitäten werden jedoch zunehmend auch genotypische Merkmale der Tumorzellen berücksichtigt. Beispiele finden sich in den aktuellen WHO-Klassifikationen der ZNS-Tumore, Leukämien/Lymphome und Weichgewebstumore. Für die Diagnostik bestimmter Tumortypen ist eine Testung auf entitätdefinierende genetische Aberrationen unabdingbar. So ermög-
SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 4/2018
7
Im Fokus: Neue Wege in der Krebsdiagnostik und personalisierten Medizin
Tabelle 1:
Anwendungen der zielgerichteten Hochdurchsatz-Sequenzierung in der molekularen Tumordiagnostik
I Tumorklassifikation I Prognoseabschätzung und Risikostratifizierung
(v.a. bei hämato-onkologischen Erkrankungen) I Identifikation von Patienten für zielgerichtete Therapien
(Standardtherapie, experimentelle Therapien ausserhalb von Zulassungsindikationen) I Identifikation von Patienten für Therapiestudien I Tumormutationslast (Ansprechen auf Immuncheckpunkt-Inhibitoren)
Tabelle 2:
Tumortypen, bei denen die Wirksamkeit einer zielgerichteten Therapie mit einer Veränderung im Tumorzellgenom assoziiert ist
Tumortyp Nicht kleinzelliges Lungenkarzinom
Kolorektales Karzinom Hochgradiges seröses Ovarialkarzinom Mammakarzinom Magenkarzinom Prostatakarzinom Melanom Gastrointestinaler Stromatumor (GIST) Chronische lymphatische Leukämie (CLL) Chronische myeloische Leukämie (CML) Akute myeloische Leukämie (AML)
Gene mit therapeutischer Bedeutung ALK, BRAF, EGFR, HER2 (ERBB2), MET, NTRK1, RET, ROS1 KRAS, NRAS BRCA1, BRCA2 HER2 (ERBB2) HER2 (ERBB2) ATM, BRCA2 BRAF, NRAS, KIT BRAF, KIT, PDGFRA TP53 ABL1 FLT3, IDH1, IDH2
licht bei histologisch undifferenzierten Sarkomen erst der Nachweis einer charakteristischen Translokation die diagnostische Einordnung. Bei der akuten myeloischen Leukämie werden klinisch und prognostisch unterschiedliche Subtypen unterschieden, die durch das Vorliegen bestimmter Mutationen (CEBPA, NPM1, FLT3 etc.) und/oder Translokationen charakterisiert sind (9). Bei myelodysplastischen Syndromen ergänzt der Nachweis bestimmter Mutationen klinische Modelle (IPSS-R) zur Risikostratifizierung (10). Die zielgerichtete Hochdurchsatz-Sequenzierung bietet sich als Untersuchungsverfahren vor allem in den Situationen an, wenn Tumorzellen auf Veränderungen in mehreren Genen untersucht werden müssen, um eine diagnostische und/oder prognostische Einordnung zur erreichen.
Identifikation von Patienten für etablierte zielgerichtete Therapien Zielgerichtete Therapien werden inzwischen bei zahlreichen Tumorentitäten als Standardtherapie eingesetzt. Die molekularpathologische Diagnostik hat hier das Ziel, genomische Veränderungen nachzuweisen, die ein Ansprechen (Sensitivität oder Resistenz) auf eine bestimmte Therapieoption vorhersagen lassen (Tabellen 1 und 2). Histologisch gleichartige nicht kleinzellige Lungenkarzinome (NSCLC) lassen sich
durch den Nachweis von Mutationen in molekulare Subtypen unterteilen, für die unterschiedliche Therapien mit in klinischen Studien belegter Wirksamkeit verfügbar sind (11, 12). Bei Tumortypen, bei denen mehrere klinisch relevante Mutationen vorliegen können, werden durch eine parallele Testung mittels Hochdurchsatz-Sequenzierung Effizienzgewinne und kürzere Bearbeitungszeiten im Vergleich zu einer sequenziellen Einzelgentestung der verschiedenen Biomarker erzielt. Dies gilt auch, falls infolge Abwesenheit von sogenannten Hotspotmutationen eine Sequenzierung des gesamten Gens (TP53, BRCA1, BRCA2 etc.) erforderlich ist.
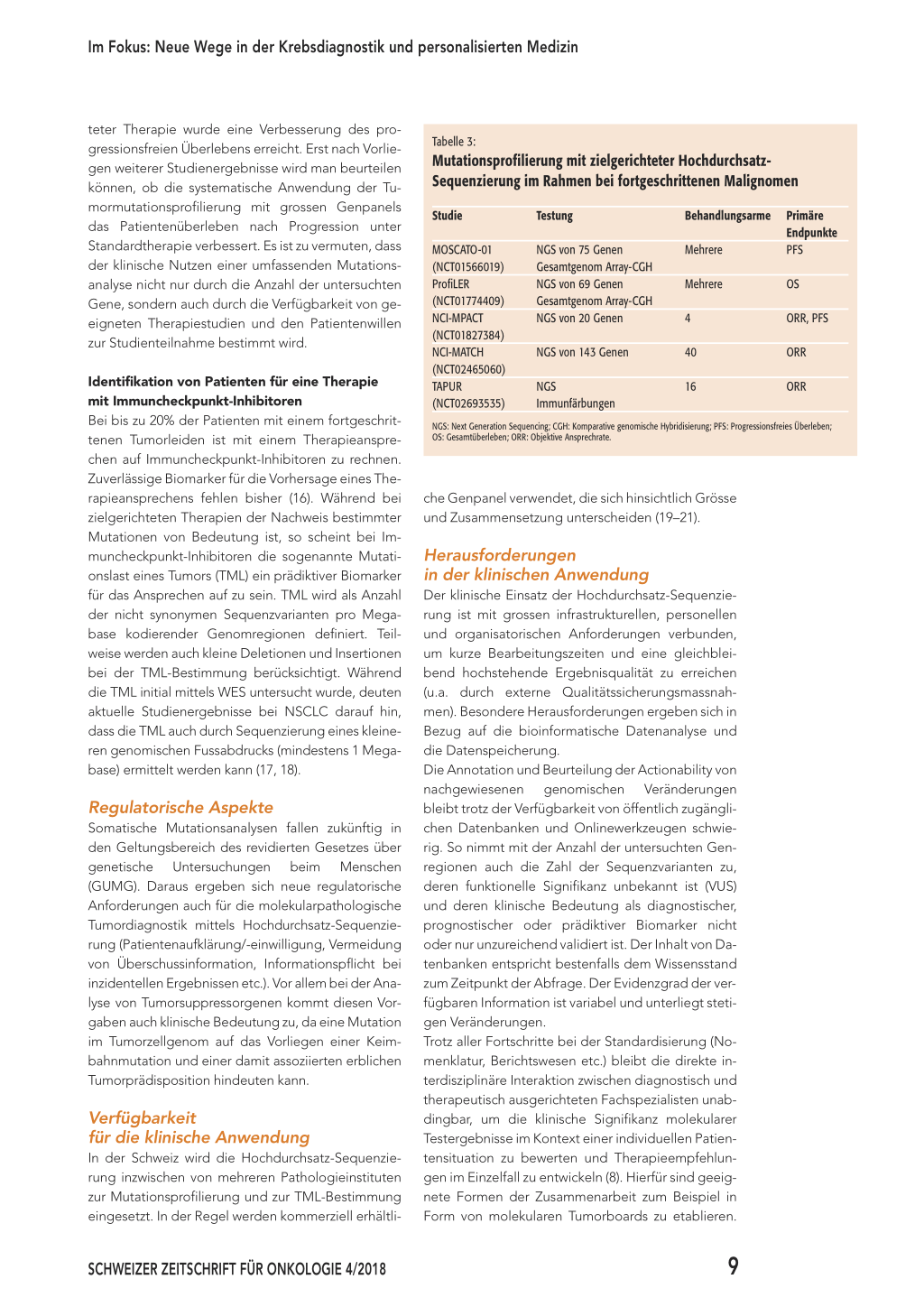
Identifikation von Patienten für experimentelle Therapien Für Patienten, bei denen eine Tumorprogression unter Standardtherapie vorliegt oder für deren Tumortyp keine wirksame Standardtherapie etabliert ist, kann der Nachweis einer Mutation die Grundlage für den Einsatz einer experimentellen Therapie sein. Ein aktuelles Beispiel sind TRK-Fusionen, die mit geringer Frequenz in verschiedenen Tumortypen vorhanden sind und durch die Entwicklung von selektiven TRK-Inhibitoren wie Larotrectinib klinische Relevanz erhalten haben (13, 14). Bei onkologischen Therapiestudien mit «Basket»oder «Umbrella»-Design erfolgt die Stratifizierung in die verschiedenen experimentellen Behandlungsarme anhand genotypischer Tumormerkmale. Das Mutationsscreening mittels zielgerichteter Hochdurchsatz-Sequenzierung ist ein effizientes Verfahren, Patienten für einen Studieneinschluss zu identifizieren. In Fallberichten ist die Verwendung der Tumormutationsprofilierung zur Identifikation von experimentellen Therapieoptionen dokumentiert. Die Machbarkeit und der klinische Nutzen einer systematischen Anwendung wird gegenwärtig in mehreren prospektiven Studien (MOSCATO-01, NCI-MPACT, NCIMATCH, TAPUR etc.) unter Verwendung von klinisch relevanten Endpunkten wie Ansprechrate und Überlebenszeit untersucht (Tabelle 3). In allen Studien kommt die Hochdurchsatz-Sequenzierung zur Anwendung. Allerdings unterscheiden sich die Studien in Bezug auf Grösse und Zusammensetzung der verwendeten Genpanel, den Einsatz zusätzlicher Untersuchungsverfahren (Array-CGH, Immunhistochemie) und die Anzahl der verfügbaren Therapiearme (bis zu 40 zugelassene oder experimentelle Wirkstoffe in der NCI-MATCH-Studie). In der MOSCATO-01-Studie konnte bei 49% der Patienten mit Progression unter Standardtherapie und erfolgreicher Testung wenigstens eine potenziell therapeutisch nutzbare Mutation identifiziert werden (15). Eine zielgerichtete Therapie war für die Hälfte der Patienten (24% der Kohorte) verfügbar und bei 33% der Patienten mit zielgerich-
8 SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 4/2018
Im Fokus: Neue Wege in der Krebsdiagnostik und personalisierten Medizin
teter Therapie wurde eine Verbesserung des progressionsfreien Überlebens erreicht. Erst nach Vorliegen weiterer Studienergebnisse wird man beurteilen können, ob die systematische Anwendung der Tumormutationsprofilierung mit grossen Genpanels das Patientenüberleben nach Progression unter Standardtherapie verbessert. Es ist zu vermuten, dass der klinische Nutzen einer umfassenden Mutationsanalyse nicht nur durch die Anzahl der untersuchten Gene, sondern auch durch die Verfügbarkeit von geeigneten Therapiestudien und den Patientenwillen zur Studienteilnahme bestimmt wird.
Identifikation von Patienten für eine Therapie mit Immuncheckpunkt-Inhibitoren Bei bis zu 20% der Patienten mit einem fortgeschrittenen Tumorleiden ist mit einem Therapieansprechen auf Immuncheckpunkt-Inhibitoren zu rechnen. Zuverlässige Biomarker für die Vorhersage eines Therapieansprechens fehlen bisher (16). Während bei zielgerichteten Therapien der Nachweis bestimmter Mutationen von Bedeutung ist, so scheint bei Immuncheckpunkt-Inhibitoren die sogenannte Mutationslast eines Tumors (TML) ein prädiktiver Biomarker für das Ansprechen auf zu sein. TML wird als Anzahl der nicht synonymen Sequenzvarianten pro Megabase kodierender Genomregionen definiert. Teilweise werden auch kleine Deletionen und Insertionen bei der TML-Bestimmung berücksichtigt. Während die TML initial mittels WES untersucht wurde, deuten aktuelle Studienergebnisse bei NSCLC darauf hin, dass die TML auch durch Sequenzierung eines kleineren genomischen Fussabdrucks (mindestens 1 Megabase) ermittelt werden kann (17, 18).
Regulatorische Aspekte
Somatische Mutationsanalysen fallen zukünftig in den Geltungsbereich des revidierten Gesetzes über genetische Untersuchungen beim Menschen (GUMG). Daraus ergeben sich neue regulatorische Anforderungen auch für die molekularpathologische Tumordiagnostik mittels Hochdurchsatz-Sequenzierung (Patientenaufklärung/-einwilligung, Vermeidung von Überschussinformation, Informationspflicht bei inzidentellen Ergebnissen etc.). Vor allem bei der Analyse von Tumorsuppressorgenen kommt diesen Vorgaben auch klinische Bedeutung zu, da eine Mutation im Tumorzellgenom auf das Vorliegen einer Keimbahnmutation und einer damit assoziierten erblichen Tumorprädisposition hindeuten kann.
Verfügbarkeit für die klinische Anwendung
In der Schweiz wird die Hochdurchsatz-Sequenzierung inzwischen von mehreren Pathologieinstituten zur Mutationsprofilierung und zur TML-Bestimmung eingesetzt. In der Regel werden kommerziell erhältli-
Tabelle 3:
Mutationsprofilierung mit zielgerichteter HochdurchsatzSequenzierung im Rahmen bei fortgeschrittenen Malignomen
Studie
MOSCATO-01 (NCT01566019) ProfiLER (NCT01774409) NCI-MPACT (NCT01827384) NCI-MATCH (NCT02465060) TAPUR (NCT02693535)
Testung
NGS von 75 Genen Gesamtgenom Array-CGH NGS von 69 Genen Gesamtgenom Array-CGH NGS von 20 Genen
NGS von 143 Genen
NGS Immunfärbungen
Behandlungsarme Mehrere
Primäre Endpunkte PFS
Mehrere
OS
4 ORR, PFS
40 ORR
16 ORR
NGS: Next Generation Sequencing; CGH: Komparative genomische Hybridisierung; PFS: Progressionsfreies Überleben; OS: Gesamtüberleben; ORR: Objektive Ansprechrate.
che Genpanel verwendet, die sich hinsichtlich Grösse und Zusammensetzung unterscheiden (19–21).
Herausforderungen in der klinischen Anwendung
Der klinische Einsatz der Hochdurchsatz-Sequenzierung ist mit grossen infrastrukturellen, personellen und organisatorischen Anforderungen verbunden, um kurze Bearbeitungszeiten und eine gleichbleibend hochstehende Ergebnisqualität zu erreichen (u.a. durch externe Qualitätssicherungsmassnahmen). Besondere Herausforderungen ergeben sich in Bezug auf die bioinformatische Datenanalyse und die Datenspeicherung. Die Annotation und Beurteilung der Actionability von nachgewiesenen genomischen Veränderungen bleibt trotz der Verfügbarkeit von öffentlich zugänglichen Datenbanken und Onlinewerkzeugen schwierig. So nimmt mit der Anzahl der untersuchten Genregionen auch die Zahl der Sequenzvarianten zu, deren funktionelle Signifikanz unbekannt ist (VUS) und deren klinische Bedeutung als diagnostischer, prognostischer oder prädiktiver Biomarker nicht oder nur unzureichend validiert ist. Der Inhalt von Datenbanken entspricht bestenfalls dem Wissensstand zum Zeitpunkt der Abfrage. Der Evidenzgrad der verfügbaren Information ist variabel und unterliegt stetigen Veränderungen. Trotz aller Fortschritte bei der Standardisierung (Nomenklatur, Berichtswesen etc.) bleibt die direkte interdisziplinäre Interaktion zwischen diagnostisch und therapeutisch ausgerichteten Fachspezialisten unabdingbar, um die klinische Signifikanz molekularer Testergebnisse im Kontext einer individuellen Patientensituation zu bewerten und Therapieempfehlungen im Einzelfall zu entwickeln (8). Hierfür sind geeignete Formen der Zusammenarbeit zum Beispiel in Form von molekularen Tumorboards zu etablieren.
SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 4/2018
9
Im Fokus: Neue Wege in der Krebsdiagnostik und personalisierten Medizin
Weitere organisatorische Herausforderungen ergeben sich bei der Umsetzung der regulatorischen Vorgaben des revidierten GUMG, die eine fachübergreifende Betrachtung der Patientenpfade erfordern.
Schlussfolgerungen und Ausblick
Die zielgerichtete Hochdurchsatz-Sequenzierung hat
sich zu einem leistungsfähigen Instrument der klini-
schen Tumordiagnostik entwickelt. Hauptanwendun-
gen sind die Testung auf genomische Alterationen,
die als Biomarker für die Diagnostik, Prognoseab-
schätzung und Vorhersage eines Therapieanspre-
chens bei onkologischen Erkrankungen verwendet
werden können. Die Indikation zur Untersuchung
grosser Genpanel zur Identifikation von experimen-
tellen Therapieoptionen sollte jedoch vorerst zurück-
haltend unter Berücksichtigung der konkreten Patien-
tensituation gestellt werden (Tumortyp, Verfügbarkeit
von Therapiestudien, Bereitschaft des Patienten zur
Teilnahme an Studien etc.). Technologische Weiter-
entwicklungen der Hochdurchsatz-Sequenzierung
betreffen die Automatisierung der Arbeitsschritte im
Labor und der bioinformatischen Datenanalyse. Mög-
liche zukünftige Anwendungen sind die Mutations-
profilierung an zirkulierender Tumor-DNA (ctDNA)
und die Einführung der WES in die klinische Tumor-
diagnostik.
I
Sabine Baumann (Erstautorin)
und Prof. Dr. med. Wolfram Jochum (Korrespondenzadresse) Institut für Pathologie Kantonsspital St. Gallen 9007 St. Gallen E-Mail: wolfram.jochum@kssg.ch
Interessenkonflikte: keine.
Merkpunkte
Quellen: 1. Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell, 2011; 144(5): 646–674. 2. Hoadley KA et al.: Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 2018; 173(2): 291–304 e6. 3. Hyman DM, Taylor BS, Baselga J: Implementing Genome-Driven Oncology. Cell, 2017; 168(4): 584–599. 4. Reuter JA, Spacek DV, Snyder MP: High-throughput sequencing technologies. Mol Cell, 2015; 58(4): 586–597. 5. den Dunnen JT et al.: HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016; 37(6): 564–569. 6. Richards S et al.: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015; 17(5): 405–424. 7. Meric-Bernstam F et al.: A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst. 2015; 107(7). 8. Li MM et al.: Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn, 2017; 19(1): 4–23. 9. Dohner H et al.: Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood, 2017; 129(4): 424–447. 10. Mufti GJ et al.: Diagnostic algorithm for lower-risk myelodysplastic syndromes. Leukemia. 2018; 32(8): 1679–1696. 11. Hanna N et al.: Systemic Therapy for Stage IV Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. 2017; 35(30): 3484–3515. 12. Jordan EJ et al.: Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017; 7(6): 596–609. 13. Amatu A, Sartore-Bianchi A, Siena S: NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open, 2016. 1(2): e000023. 14. Drilon A et al.: Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med. 2018; 378(8): 731–739. 15. Massard C et al.: High-Throughput Genomics and Clinical Outcome in Hardto-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017; 7(6): 586–595. 16. Gibney GT, Weiner LM, M.B. Atkins MB: Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016; 17(12): e542–e551. 17. Rizvi H et al.: Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J Clin Oncol, 2018; 36(7): 633–641. 18. Hellmann MD et al.: Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N Engl J Med. 2018; 378(22): 2093–2104. 19. Frampton GM et al.: Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013; 31(11): 1023–1031. 20. Hovelson HD et al.: Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia, 2015. 17(4): 385–399. 21. Lih CJ et al.: Analytical Validation of the Next-Generation Sequencing Assay for a Nationwide Signal-Finding Clinical Trial: Molecular Analysis for Therapy Choice Clinical Trial. J Mol Diagn. 2017; 19(2): 313-327.
I Veränderungen im Tumorzellgenom können als diagnostische und prognostische Biomarker sowie zur Vorhersage eines Therapieansprechens bei onkologischen Erkrankungen verwendet werden.
I Die Technologie der Hochdurchsatz-Sequenzierung ermöglicht die parallele Analyse auf Mutationen in mehreren Genen/Genomabschnitten.
I Die zielgerichtete Hochdurchsatz-Sequenzierung wird zur selektiven Untersuchung von für die personalisierte Krebstherapie relevanten Genen/Genomabschnitten eingesetzt und ist für die klinische Anwendung in Pathologie-Instituten verfügbar.
10 SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 4/2018