

Transkript
FORTBILDUNG
Galaktose als einzige Energiequelle im Gehirn
Hirnfunktion: Galaktose und die Konsequenzen für die Kognition
Glukose ist die einzige Energiequelle des Gehirns. Somit hat jegliche Einschränkung der zerebralen Glukoseversorgung direkte Auswirkungen auf die Hirnfunktionen. Eine wesentliche Rolle spielt dabei der Insulinrezeptor, sein Defekt schränkt insbesondere kognitive Leistungen ein. Galaktose, der 4-epimere Zucker zur Glukose, wird Insulinrezeptor-unabhängig von Nervenzellen aufgenommen und könnte somit eine ernährungsmedizinische Möglichkeit sein, kognitive Leistungen durch Ausgleich des Energiebedarfs unter diesen Mangelbedingungen zu normalisieren.
Christoph C. Geilen Werner Reutter †
von Christoph C. Geilen und Werner Reutter*
Die zerebrale Energieversorgung
V iele körperliche und geistige Funktionen zollen dem Alter ihren Tribut. Gerade die verminderte Durchblutung von Organen und die dadurch eingeschränkte Versorgung mit Sauerstoff und Glukose tragen dazu bei. Glukose ist für das Gehirn die einzige Quelle für die Gewinnung von Energie und für die Aufrechterhaltung des Baustoffwechsels notwendig. Andere Organe wie Leber, Muskulatur oder Fettgewebe können ausser Glukose auch Aminosäuren oder Fettsäuren verwerten. Das Gehirn ist hierzu nicht in der Lage. Es benötigt zirka 150 g Glukose pro Tag. Im Gesamtblut sind jedoch nur 5 g vorrätig. Demzufolge ist das Gehirn auf die laufende Zufuhr angewiesen. Somit führt eine eingeschränkte Glukoseversorgung des Gehirns unweigerlich zu Funktionseinschränkungen. Auch im Gehirn wird die Glukoseaufnahme teilweise über den Insulinrezeptor vermittelt. Eine besonders hohe Insulinrezeptordichte findet sich im Hippocampus und im Cortex, was darauf hindeutet, dass die insulinvermittelte Glukoseaufnahme besonders für Lernprozesse und Gedächtnisleistungen wichtig ist (1).
Galaktose, die insulinunabhängige Energiequelle Seit Kurzem sind bis anhin nicht bekannte positive Eigenschaften von Galaktose für den Energiestoffwechsel des Gehirns bekannt. D-Galaktose unterscheidet sich von der D-Glukose (Traubenzucker) nur durch die Stellung einer OH-Gruppe am C4-Atom des Moleküls; die
* In memoriam meines Co-Autors Prof. Dr. med. Werner Reutter, der während der Verfassung dieses Manuskripts unerwartet verstorben ist.
Summenformel ist identisch (Abbildung 1). Dieser stereochemische Unterschied hat weitreichende Auswirkungen für den Stoffwechsel dieser beiden Zucker und kann ernährungsbiologisch genutzt werden. Beim Menschen ist D-Galaktose wesentlicher Bestandteil von Glykoproteinen und Glykolipiden. Glykoproteine und Glykolipide (Glykokonjugate) sind Bausteine von Plasmamembranen, die als Glykokalyx alle Zellen umhüllen. Sie bilden eine Schutzbarriere der Zellen, vermitteln aber auch den Kontakt zum umgebenden Milieu, zur extrazellulären Matrix und zu Nachbarzellen. Freie D-Galaktose ist ohne exogene Zufuhr nicht im Blut nachweisbar. In Nahrungsmitteln kommt freie D-Galaktose kaum vor. Gebundene D-Galaktose hingegen findet sich in verschiedenen Sacchariden. Das einfachste galaktosehaltige Saccharid ist das Disaccharid Laktose (Milchzucker). Es besteht aus einem Glukose- und einem Galaktosemolekül. Laktose kommt vorwiegend in Milchprodukten vor. Im Zellstoffwechsel können Galaktose und Glukose über die Stufen der aktivierten Monosaccharide (UDPGlukose und UDP-Galaktose) ineinander überführt werden (Abbildung 2). Die Metabolisierung exogen zugeführter Galaktose aus der Nahrung erfolgt über ihre Phosphorylierung mithilfe von Adenosintriphosphat (ATP) und der hochspezifischen Galaktokinase und der anschliessenden Aktivierung zu UDP-Galaktose durch die Uridylyltransferase und UDP-Glukose. Dieses Enzym ist bei der hereditären Galaktosämie defekt. Die Aufrechterhaltung einer physiologischen Glukosekonzentration im Blut und in den Zellen ist eine sehr wichtige Aufgabe des Stoffwechsels. Dafür sorgt das Hormon Insulin. Insulin stimuliert den Einstrom von Glukose in die Zellen der Organe (mit Ausnahme der Leber). Um seinen Befehl an Zellen zu vermitteln, benötigt Insulin den Insulinrezeptor, da es als Peptidhormon nicht in die Zellen eindringen kann. Hat Insulin an seinen
&30 1/2017
PSYCHIATRIE NEUROLOGIE
FORTBILDUNG
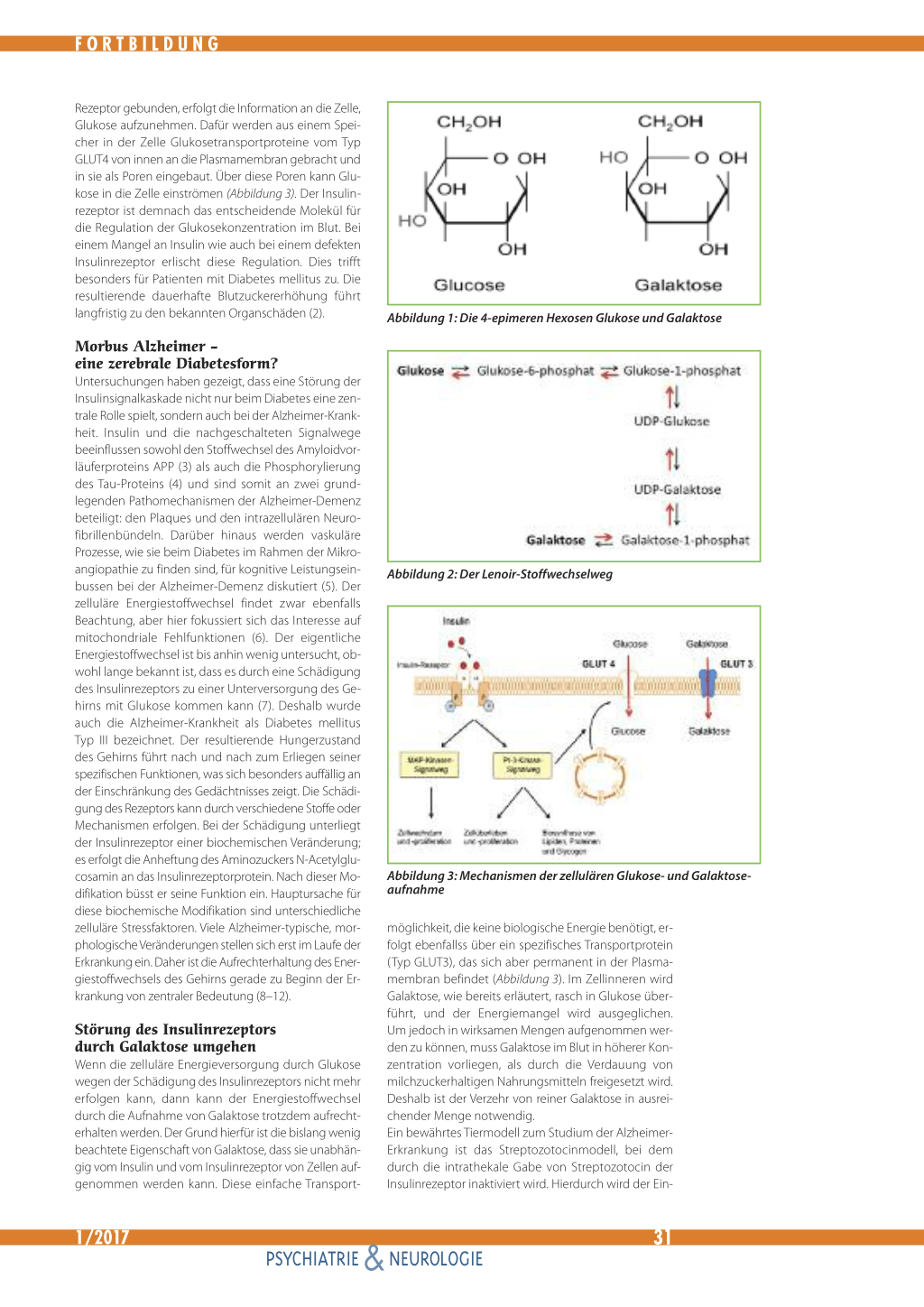
Rezeptor gebunden, erfolgt die Information an die Zelle, Glukose aufzunehmen. Dafür werden aus einem Speicher in der Zelle Glukosetransportproteine vom Typ GLUT4 von innen an die Plasmamembran gebracht und in sie als Poren eingebaut. Über diese Poren kann Glukose in die Zelle einströmen (Abbildung 3). Der Insulinrezeptor ist demnach das entscheidende Molekül für die Regulation der Glukosekonzentration im Blut. Bei einem Mangel an Insulin wie auch bei einem defekten Insulinrezeptor erlischt diese Regulation. Dies trifft besonders für Patienten mit Diabetes mellitus zu. Die resultierende dauerhafte Blutzuckererhöhung führt langfristig zu den bekannten Organschäden (2).
Morbus Alzheimer – eine zerebrale Diabetesform? Untersuchungen haben gezeigt, dass eine Störung der Insulinsignalkaskade nicht nur beim Diabetes eine zentrale Rolle spielt, sondern auch bei der Alzheimer-Krankheit. Insulin und die nachgeschalteten Signalwege beeinflussen sowohl den Stoffwechsel des Amyloidvorläuferproteins APP (3) als auch die Phosphorylierung des Tau-Proteins (4) und sind somit an zwei grundlegenden Pathomechanismen der Alzheimer-Demenz beteiligt: den Plaques und den intrazellulären Neurofibrillenbündeln. Darüber hinaus werden vaskuläre Prozesse, wie sie beim Diabetes im Rahmen der Mikroangiopathie zu finden sind, für kognitive Leistungseinbussen bei der Alzheimer-Demenz diskutiert (5). Der zelluläre Energiestoffwechsel findet zwar ebenfalls Beachtung, aber hier fokussiert sich das Interesse auf mitochondriale Fehlfunktionen (6). Der eigentliche Energiestoffwechsel ist bis anhin wenig untersucht, obwohl lange bekannt ist, dass es durch eine Schädigung des Insulinrezeptors zu einer Unterversorgung des Gehirns mit Glukose kommen kann (7). Deshalb wurde auch die Alzheimer-Krankheit als Diabetes mellitus Typ III bezeichnet. Der resultierende Hungerzustand des Gehirns führt nach und nach zum Erliegen seiner spezifischen Funktionen, was sich besonders auffällig an der Einschränkung des Gedächtnisses zeigt. Die Schädigung des Rezeptors kann durch verschiedene Stoffe oder Mechanismen erfolgen. Bei der Schädigung unterliegt der Insulinrezeptor einer biochemischen Veränderung; es erfolgt die Anheftung des Aminozuckers N-Acetylglucosamin an das Insulinrezeptorprotein. Nach dieser Modifikation büsst er seine Funktion ein. Hauptursache für diese biochemische Modifikation sind unterschiedliche zelluläre Stressfaktoren. Viele Alzheimer-typische, morphologische Veränderungen stellen sich erst im Laufe der Erkrankung ein. Daher ist die Aufrechterhaltung des Energiestoffwechsels des Gehirns gerade zu Beginn der Erkrankung von zentraler Bedeutung (8–12).
Störung des Insulinrezeptors durch Galaktose umgehen Wenn die zelluläre Energieversorgung durch Glukose wegen der Schädigung des Insulinrezeptors nicht mehr erfolgen kann, dann kann der Energiestoffwechsel durch die Aufnahme von Galaktose trotzdem aufrechterhalten werden. Der Grund hierfür ist die bislang wenig beachtete Eigenschaft von Galaktose, dass sie unabhängig vom Insulin und vom Insulinrezeptor von Zellen aufgenommen werden kann. Diese einfache Transport-
Abbildung 1: Die 4-epimeren Hexosen Glukose und Galaktose
Abbildung 2: Der Lenoir-Stoffwechselweg
Abbildung 3: Mechanismen der zellulären Glukose- und Galaktoseaufnahme möglichkeit, die keine biologische Energie benötigt, erfolgt ebenfallss über ein spezifisches Transportprotein (Typ GLUT3), das sich aber permanent in der Plasmamembran befindet (Abbildung 3). Im Zellinneren wird Galaktose, wie bereits erläutert, rasch in Glukose überführt, und der Energiemangel wird ausgeglichen. Um jedoch in wirksamen Mengen aufgenommen werden zu können, muss Galaktose im Blut in höherer Konzentration vorliegen, als durch die Verdauung von milchzuckerhaltigen Nahrungsmitteln freigesetzt wird. Deshalb ist der Verzehr von reiner Galaktose in ausreichender Menge notwendig. Ein bewährtes Tiermodell zum Studium der AlzheimerErkrankung ist das Streptozotocinmodell, bei dem durch die intrathekale Gabe von Streptozotocin der Insulinrezeptor inaktiviert wird. Hierdurch wird der Ein-
1/2017
PSYCHIATRIE & NEUROLOGIE
31
FORTBILDUNG
Merkpunkte:
G Glukose ist das einzige Energiesubstrat des Gehirns. G Störungen der Glukoseversorgung führen zu Störungen der Hirnfunktionen. G Beim Morbus Alzheimer und bei hepatischen Enzephalopathien wird häufig ein
Defekt in der Insulinrezeptor-Signalkaskade gefunden. G Galaktose wird unabhängig vom Insulinrezeptor aufgenommen und intrazellu-
lär in Glukose umgewandelt.
strom von Glukose über das Insulinrezeptorabhängige Transportprotein GLUT4 in die Hirnzellen nachhaltig gehemmt. Das Gehirn wird nicht mehr ausreichend mit dem essenziellen Energiesubstrat Glukose versorgt. Es kommt zu neurodegenerativen Veränderungen, und die behandelten Tiere büssen messbar ihre Gedächtnisleistung ein. Bemerkenswerterweise konnte durch Zugabe von Galaktose in das Trinkwasser nicht nur das zelluläre Energiedefizit behoben, sondern auch die kognitiven Leistungen der Versuchstiere wesentlich verbessert werden (13).
Fazit
Diabetes und Morbus Alzheimer weisen eine Reihe epi-
demiologischer und pathophysiologischer Gemeinsam-
keiten auf. Hierbei steht besonders Insulin und seine
nachgeschalteten Signal- und Stoffwechselwege im
Fokus des Interesses.
G
Korrespondenzadresse: Prof. Dr. med. Dr. rer. nat. Christoph C. Geilen
Fakultät für Humanwissenschaften Medical School Hamburg Am Kaiserkai 1 D-20457 Hamburg
E-Mail: christoph.geilen@medicalschool-hamburg.de
Interessenkonflikt:
Prof. Dr. Dr. Christoph C. Geilen ist geschäftsfuḧ render Gesellschafter von Glycana UG (haftungsbeschränkt), einem Biotechnologieunternehmen auf dem Gebiet der Glycobiologie, das unter anderem Galaktose als Nahrungsergänzungsmittel vertreibt.
Literatur:
1. Simpson IA, Carruthers A, Vannucci SJ: Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab. 2007; 27(11): 1766–1791.
2. Roser M, Josic D, Kontou M, Mosetter K, Maurer P, Reutter W: Metabolism of gaLaktose in the brain and liver of rats and its conversion into glutamate and other amino acids. J Neural Transm (Vienna). 2009; 116(2): 131–139.
3. Solano DC, Sironi M, Bonfini C, Solerte SB, Govoni S, Racchi M: Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB j. 2000; 14: 1015–1022.
4. El Khoury NB, Gratuze M, Papon MA, Bretteville A, Planel E: Insulin dysfunction and Tau pathology. Front Cell Neurosci. 2014; 8: 22.
5. Kloppenborg RP, van den Berg E, Kappelle LJ, Biessels GJ: Diabetes and other vascular risk factors for dementia: which factor matters most? A systematic review. Eur J Pharmacol. 2008; 585: 97–108.
6. Moreira PI, Santos MS, Seica R, Oliveira CR: Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J Neurol Sci. 2007; 257: 206–214.
7. Blum-Degen D, Frölich L, Hoyer S, Riederer P: Altered regulation of brain Glukose metabolism as a cause of neurodegenerative disorders? J Neural Transm Suppl. 195; 46: 139–147.
8. Hoyer S, Oesterreich K, Wagner O: Glukose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J Neurol. 1988; 235(3): 143–148.
9. Hoyer S: Abnormalities of Glukose metabolism in Alzheimer’s disease. Ann N Y Acad Sci. 1991; 640: 53–58.
10. Hoyer S: Glukose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 2004; 490(1–3): 115–125.
11. Morgen K, Fröhlich L: The metabolism hypothesis of Alzheimer’s disease: from the concept of central insulin resistance and associated consequences to insulin therapy. J Neural Transm 2015; 122: 499– 504.
12. Freude S, Schilbach K, Schubert M: The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: from modelorganisms to human disease. Current Alzheimer Research, 2009; 6(3): 213–223.
13. Salkovic-Petrisic M, Osmanovic-Barilar J, Knezovic A, Hoyer S, Mosetter K, Reutter W: Long-term oral gaLaktose treatment prevents cognitive deficits in male Wistar rats treated intracerebroventricularly with streptozotocin. Neuropharmacology. 2014; 77: 68–80.
&32 1/2017
PSYCHIATRIE NEUROLOGIE