

Transkript
SCHWERPUNKT
Unklares Fieber
Diagnostische Schwierigkeiten bei inkomplettem Kawasaki-Syndrom
Das Kawasaki-Syndrom ist eine der häufigsten Vaskulitiden im Kindesalter. Das rechtzeitige Erkennen der Symptome ist wichtig, da sich bei unbehandelten Patienten in 15 bis 25 Prozent der Fälle Koronaraneurysmen entwickeln. Das Kawasaki-Syndrom hat damit das rheumatische Fieber als häufigste Ursache von erworbenen Herzerkrankungen abgelöst. Es tritt typischerweise im Kleinkindalter auf, am häufigsten zwischen dem ersten und zweiten Lebensjahr.
Von Elea Galiart, Birgit Donner und Andreas Wörner
Unbehandelte Patienten entwickeln in 15 bis 25 Prozent der Fälle Koronaraneurysmen.
Ein bisher gesundes, drei Monate altes Mädchen wurde uns mit Fieber seit dem Vortag bis maximal 39° C ohne adäquaten Fokus und mit erhöhten Entzündungswerten zugewiesen.
Es erfolgte eine Standarddiagnostik mit differenziertem Blutbild, CRP und Blutkultur. Zudem wurde eine Lumbalpunktion, ein Urinstatus mittels Katheterurin und ein Thoraxröntgen durchgeführt. Dabei zeigten sich bis auf ein erhöhtes CRP von 49 mg/l und eine Leukozytose von 23,5 × 109/l (Normwert 6–17,5 × 109/l), davon 67,4 Prozent Neutrophile, unauffällige Befunde. Aufgrund des jungen Alters und des fehlenden Fokus wurde eine empirische antibiotische Therapie mit Ceftriaxon 100 mg/kg KG/Tag gestartet. Am Folgetag fiel eine leichte Rhinitis auf, im Nasopharyngealsekret konnte Rhinovirus/Enterovirus nachgewiesen werde. Eine zudem aufgetretene seröse Konjunktivis und ein ausgeprägtes makulopapulöses, im Verlauf konfluierendes Exanthem wurde im Rahmen der Virusinfektion interpretiert. Die antibiotische Therapie konnte nach Erhalt der negativen Liquor- und Blutkultur nach insgesamt 72 Stunden gestoppt werden. Bei unverändertem CRP, ordentlichem Allgemeinzustand und gutem Trinkverhalten erfolgte, trotz weiterhin intermittierend febrilen Temperaturen, nach fünf Tagen Hospitalisation die Entlassung nach Hause. Zwei Tage nach Entlassung wurde von den Eltern eine persistierende Unruhe des Kindes mit wiederholtem Überstrecken des Kopfes beobachtet, zudem traten weiterhin intermittierend febrile Temperaturen bis maximal 39 °C auf, sodass die erneute Vorstellung auf der Notfallstation erfolgte. Klinisch präsentierte sich das Mädchen in leicht reduziertem Allgemeinzustand und war irritabel bei wei-
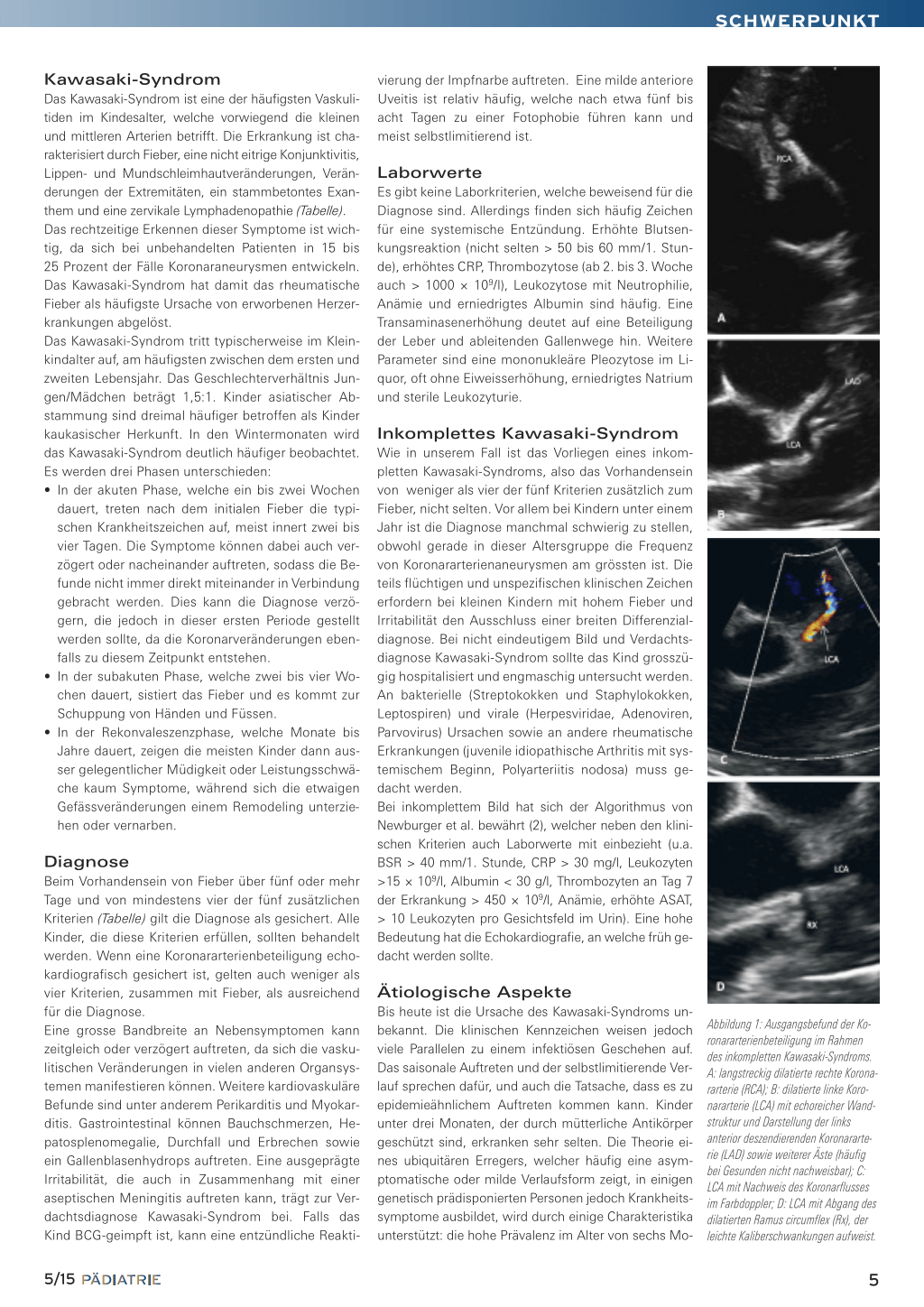
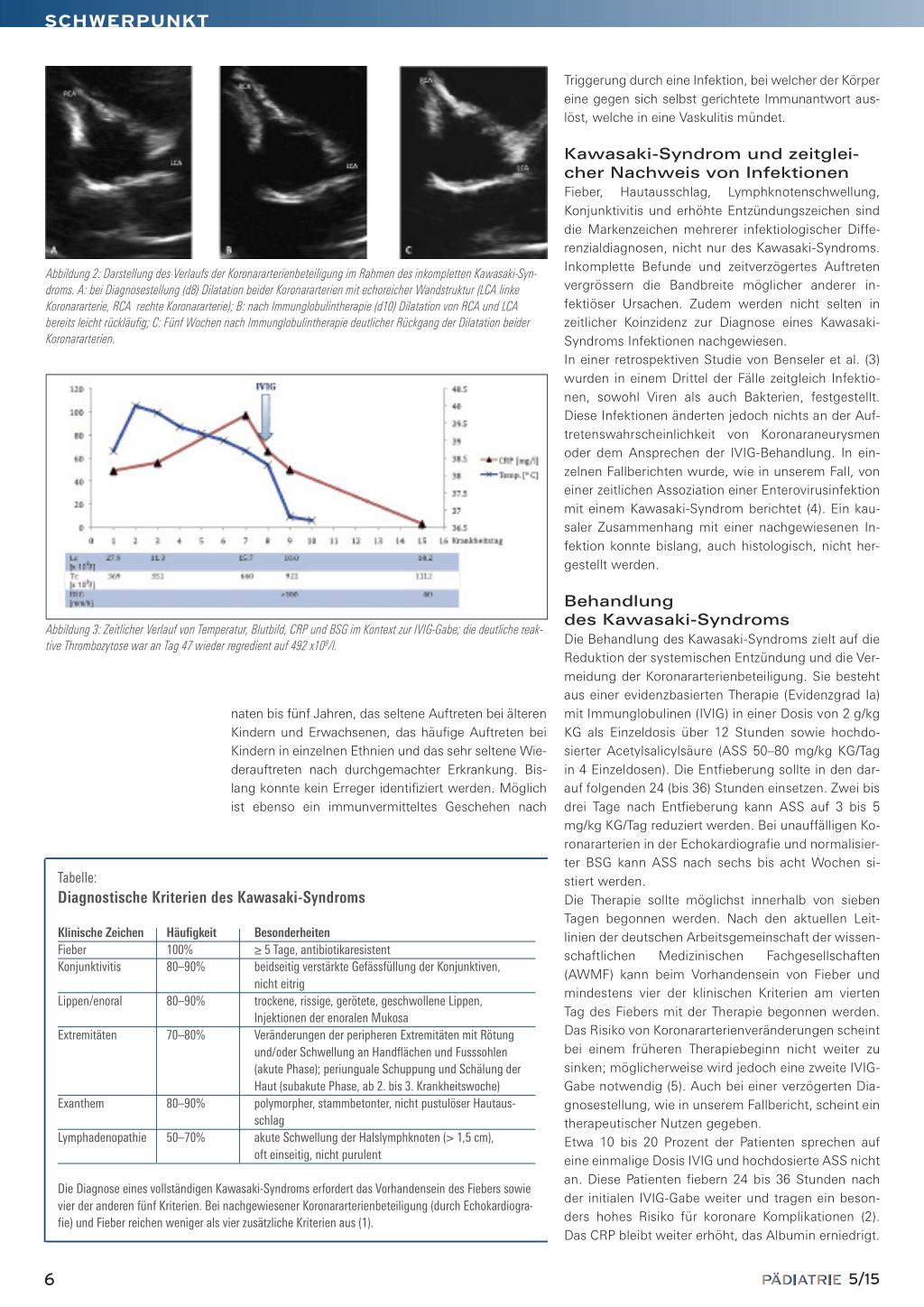
cher und nicht vorgewölbter Fontanelle. Das Exanthem war komplett regredient. Zeichen für eine Konjunktivitis bestanden nicht mehr. Eine Lumbalpunktion war vergeblich, weshalb bei ordentlichem Allgemeinzustand auf eine Punktion verzichtet wurde. Im Labor fiel ein erneuter CRP-Anstieg auf 97 mg/l bei normwertigen Leukozyten (15,7 × 109/l) und eine Thrombozytose von 660 × 109/l (Normwert: 150– 450 × 109/l) auf. Die BSG war mit > 100 mm/1. Stunde deutlich erhöht. In Zusammenschau der Befunde mit prolongiertem Fieber, Status nach nicht eitriger Konjunktivitis, makulopapulösem Exanthem, erneutem CRP-Anstieg, erhöhter BSG und im Verlauf Auftreten eine Thrombozytose wurde differenzialdiagnostisch an ein inkomplettes Kawasaki-Syndrom gedacht. In der Echokardiografie liess sich der Verdacht erhärten: Es zeigten sich im Bereich der linken und rechten Koronararterie langstreckig echoreiche Wandstrukturen und Kaliberunregelmässigkeiten mit dilatierten Gefässen bis maximal 3 mm, zudem bestand ein geringer, zirkulärer Perikarderguss (Abbildung 1). Es wurde umgehend eine Therapie mit Immunglobulinen i.v. (2 g/kg KG über 12 Stunden) und hochdosierter Acetylsalicylsäure p.o. (66 mg/kg KG/Tag in 4 Einzeldosen) initiiert. Nach Immunglobulingabe präsentierte sich das Mädchen in deutlich gebessertem Allgemeinzustand, war nicht mehr irritabel und blieb ab dem Folgetag afebril. Die Therapie mit Acetylsalicylsäure wurde 48 Stunden nach Entfieberung auf 3 mg/kg KG/Tag reduziert. In der ambulanten Nachkontrolle, sieben Tage nach Immunglobulingabe, zeigten sich bereits eine leicht rückläufige Koronararterienbeteiligung und ein vollständiger Rückgang des Perikardergusses (Abbildung 2). Zudem fiel eine leichte Hautschuppung der Finger auf. In Abbildung 3 ist der Verlauf der Laborwerte dargestellt.
4 5/15
SCHWERPUNKT
Kawasaki-Syndrom
Das Kawasaki-Syndrom ist eine der häufigsten Vaskulitiden im Kindesalter, welche vorwiegend die kleinen und mittleren Arterien betrifft. Die Erkrankung ist charakterisiert durch Fieber, eine nicht eitrige Konjunktivitis, Lippen- und Mundschleimhautveränderungen, Veränderungen der Extremitäten, ein stammbetontes Exanthem und eine zervikale Lymphadenopathie (Tabelle). Das rechtzeitige Erkennen dieser Symptome ist wichtig, da sich bei unbehandelten Patienten in 15 bis 25 Prozent der Fälle Koronaraneurysmen entwickeln. Das Kawasaki-Syndrom hat damit das rheumatische Fieber als häufigste Ursache von erworbenen Herzerkrankungen abgelöst. Das Kawasaki-Syndrom tritt typischerweise im Kleinkindalter auf, am häufigsten zwischen dem ersten und zweiten Lebensjahr. Das Geschlechterverhältnis Jungen/Mädchen beträgt 1,5:1. Kinder asiatischer Abstammung sind dreimal häufiger betroffen als Kinder kaukasischer Herkunft. In den Wintermonaten wird das Kawasaki-Syndrom deutlich häufiger beobachtet. Es werden drei Phasen unterschieden: • In der akuten Phase, welche ein bis zwei Wochen
dauert, treten nach dem initialen Fieber die typischen Krankheitszeichen auf, meist innert zwei bis vier Tagen. Die Symptome können dabei auch verzögert oder nacheinander auftreten, sodass die Befunde nicht immer direkt miteinander in Verbindung gebracht werden. Dies kann die Diagnose verzögern, die jedoch in dieser ersten Periode gestellt werden sollte, da die Koronarveränderungen ebenfalls zu diesem Zeitpunkt entstehen. • In der subakuten Phase, welche zwei bis vier Wochen dauert, sistiert das Fieber und es kommt zur Schuppung von Händen und Füssen. • In der Rekonvaleszenzphase, welche Monate bis Jahre dauert, zeigen die meisten Kinder dann ausser gelegentlicher Müdigkeit oder Leistungsschwäche kaum Symptome, während sich die etwaigen Gefässveränderungen einem Remodeling unterziehen oder vernarben.
Diagnose
Beim Vorhandensein von Fieber über fünf oder mehr Tage und von mindestens vier der fünf zusätzlichen Kriterien (Tabelle) gilt die Diagnose als gesichert. Alle Kinder, die diese Kriterien erfüllen, sollten behandelt werden. Wenn eine Koronararterienbeteiligung echokardiografisch gesichert ist, gelten auch weniger als vier Kriterien, zusammen mit Fieber, als ausreichend für die Diagnose. Eine grosse Bandbreite an Nebensymptomen kann zeitgleich oder verzögert auftreten, da sich die vaskulitischen Veränderungen in vielen anderen Organsystemen manifestieren können. Weitere kardiovaskuläre Befunde sind unter anderem Perikarditis und Myokarditis. Gastrointestinal können Bauchschmerzen, Hepatosplenomegalie, Durchfall und Erbrechen sowie ein Gallenblasenhydrops auftreten. Eine ausgeprägte Irritabilität, die auch in Zusammenhang mit einer aseptischen Meningitis auftreten kann, trägt zur Verdachtsdiagnose Kawasaki-Syndrom bei. Falls das Kind BCG-geimpft ist, kann eine entzündliche Reakti-
vierung der Impfnarbe auftreten. Eine milde anteriore Uveitis ist relativ häufig, welche nach etwa fünf bis acht Tagen zu einer Fotophobie führen kann und meist selbstlimitierend ist.
Laborwerte
Es gibt keine Laborkriterien, welche beweisend für die Diagnose sind. Allerdings finden sich häufig Zeichen für eine systemische Entzündung. Erhöhte Blutsenkungsreaktion (nicht selten > 50 bis 60 mm/1. Stunde), erhöhtes CRP, Thrombozytose (ab 2. bis 3. Woche auch > 1000 × 109/l), Leukozytose mit Neutrophilie, Anämie und erniedrigtes Albumin sind häufig. Eine Transaminasenerhöhung deutet auf eine Beteiligung der Leber und ableitenden Gallenwege hin. Weitere Parameter sind eine mononukleäre Pleozytose im Liquor, oft ohne Eiweisserhöhung, erniedrigtes Natrium und sterile Leukozyturie.
Inkomplettes Kawasaki-Syndrom
Wie in unserem Fall ist das Vorliegen eines inkompletten Kawasaki-Syndroms, also das Vorhandensein von weniger als vier der fünf Kriterien zusätzlich zum Fieber, nicht selten. Vor allem bei Kindern unter einem Jahr ist die Diagnose manchmal schwierig zu stellen, obwohl gerade in dieser Altersgruppe die Frequenz von Koronararterienaneurysmen am grössten ist. Die teils flüchtigen und unspezifischen klinischen Zeichen erfordern bei kleinen Kindern mit hohem Fieber und Irritabilität den Ausschluss einer breiten Differenzialdiagnose. Bei nicht eindeutigem Bild und Verdachtsdiagnose Kawasaki-Syndrom sollte das Kind grosszügig hospitalisiert und engmaschig untersucht werden. An bakterielle (Streptokokken und Staphylokokken, Leptospiren) und virale (Herpesviridae, Adenoviren, Parvovirus) Ursachen sowie an andere rheumatische Erkrankungen (juvenile idiopathische Arthritis mit systemischem Beginn, Polyarteriitis nodosa) muss gedacht werden. Bei inkomplettem Bild hat sich der Algorithmus von Newburger et al. bewährt (2), welcher neben den klinischen Kriterien auch Laborwerte mit einbezieht (u.a. BSR > 40 mm/1. Stunde, CRP > 30 mg/l, Leukozyten >15 × 109/l, Albumin < 30 g/l, Thrombozyten an Tag 7 der Erkrankung > 450 × 109/l, Anämie, erhöhte ASAT, > 10 Leukozyten pro Gesichtsfeld im Urin). Eine hohe Bedeutung hat die Echokardiografie, an welche früh gedacht werden sollte.
Ätiologische Aspekte
Bis heute ist die Ursache des Kawasaki-Syndroms unbekannt. Die klinischen Kennzeichen weisen jedoch viele Parallelen zu einem infektiösen Geschehen auf. Das saisonale Auftreten und der selbstlimitierende Verlauf sprechen dafür, und auch die Tatsache, dass es zu epidemieähnlichem Auftreten kommen kann. Kinder unter drei Monaten, der durch mütterliche Antikörper geschützt sind, erkranken sehr selten. Die Theorie eines ubiquitären Erregers, welcher häufig eine asymptomatische oder milde Verlaufsform zeigt, in einigen genetisch prädisponierten Personen jedoch Krankheitssymptome ausbildet, wird durch einige Charakteristika unterstützt: die hohe Prävalenz im Alter von sechs Mo-
Abbildung 1: Ausgangsbefund der Koronararterienbeteiligung im Rahmen des inkompletten Kawasaki-Syndroms. A: langstreckig dilatierte rechte Koronararterie (RCA); B: dilatierte linke Koronararterie (LCA) mit echoreicher Wandstruktur und Darstellung der links anterior deszendierenden Koronararterie (LAD) sowie weiterer Äste (häufig bei Gesunden nicht nachweisbar); C: LCA mit Nachweis des Koronarflusses im Farbdoppler; D: LCA mit Abgang des dilatierten Ramus circumflex (Rx), der leichte Kaliberschwankungen aufweist.
5/15
5
SCHWERPUNKT
Triggerung durch eine Infektion, bei welcher der Körper eine gegen sich selbst gerichtete Immunantwort auslöst, welche in eine Vaskulitis mündet.
Abbildung 2: Darstellung des Verlaufs der Koronararterienbeteiligung im Rahmen des inkompletten Kawasaki-Syndroms. A: bei Diagnosestellung (d8) Dilatation beider Koronararterien mit echoreicher Wandstruktur (LCA linke Koronararterie, RCA rechte Koronararterie); B: nach Immunglobulintherapie (d10) Dilatation von RCA und LCA bereits leicht rückläufig; C: Fünf Wochen nach Immunglobulintherapie deutlicher Rückgang der Dilatation beider Koronararterien.
Kawasaki-Syndrom und zeitgleicher Nachweis von Infektionen
Fieber, Hautausschlag, Lymphknotenschwellung, Konjunktivitis und erhöhte Entzündungszeichen sind die Markenzeichen mehrerer infektiologischer Differenzialdiagnosen, nicht nur des Kawasaki-Syndroms. Inkomplette Befunde und zeitverzögertes Auftreten vergrössern die Bandbreite möglicher anderer infektiöser Ursachen. Zudem werden nicht selten in zeitlicher Koinzidenz zur Diagnose eines KawasakiSyndroms Infektionen nachgewiesen. In einer retrospektiven Studie von Benseler et al. (3) wurden in einem Drittel der Fälle zeitgleich Infektionen, sowohl Viren als auch Bakterien, festgestellt. Diese Infektionen änderten jedoch nichts an der Auftretenswahrscheinlichkeit von Koronaraneurysmen oder dem Ansprechen der IVIG-Behandlung. In einzelnen Fallberichten wurde, wie in unserem Fall, von einer zeitlichen Assoziation einer Enterovirusinfektion mit einem Kawasaki-Syndrom berichtet (4). Ein kausaler Zusammenhang mit einer nachgewiesenen Infektion konnte bislang, auch histologisch, nicht hergestellt werden.
Abbildung 3: Zeitlicher Verlauf von Temperatur, Blutbild, CRP und BSG im Kontext zur IVIG-Gabe; die deutliche reaktive Thrombozytose war an Tag 47 wieder regredient auf 492 x109/l.
naten bis fünf Jahren, das seltene Auftreten bei älteren Kindern und Erwachsenen, das häufige Auftreten bei Kindern in einzelnen Ethnien und das sehr seltene Wiederauftreten nach durchgemachter Erkrankung. Bislang konnte kein Erreger identifiziert werden. Möglich ist ebenso ein immunvermitteltes Geschehen nach
Tabelle: Diagnostische Kriterien des Kawasaki-Syndroms
Klinische Zeichen Fieber Konjunktivitis
Häufigkeit 100% 80–90%
Lippen/enoral
80–90%
Extremitäten
70–80%
Exanthem
80–90%
Lymphadenopathie 50–70%
Besonderheiten ≥ 5 Tage, antibiotikaresistent beidseitig verstärkte Gefässfüllung der Konjunktiven, nicht eitrig trockene, rissige, gerötete, geschwollene Lippen, Injektionen der enoralen Mukosa Veränderungen der peripheren Extremitäten mit Rötung und/oder Schwellung an Handflächen und Fusssohlen (akute Phase); periunguale Schuppung und Schälung der Haut (subakute Phase, ab 2. bis 3. Krankheitswoche) polymorpher, stammbetonter, nicht pustulöser Hautausschlag akute Schwellung der Halslymphknoten (> 1,5 cm), oft einseitig, nicht purulent
Die Diagnose eines vollständigen Kawasaki-Syndroms erfordert das Vorhandensein des Fiebers sowie vier der anderen fünf Kriterien. Bei nachgewiesener Koronararterienbeteiligung (durch Echokardiografie) und Fieber reichen weniger als vier zusätzliche Kriterien aus (1).
Behandlung des Kawasaki-Syndroms
Die Behandlung des Kawasaki-Syndroms zielt auf die Reduktion der systemischen Entzündung und die Vermeidung der Koronararterienbeteiligung. Sie besteht aus einer evidenzbasierten Therapie (Evidenzgrad Ia) mit Immunglobulinen (IVIG) in einer Dosis von 2 g/kg KG als Einzeldosis über 12 Stunden sowie hochdosierter Acetylsalicylsäure (ASS 50–80 mg/kg KG/Tag in 4 Einzeldosen). Die Entfieberung sollte in den darauf folgenden 24 (bis 36) Stunden einsetzen. Zwei bis drei Tage nach Entfieberung kann ASS auf 3 bis 5 mg/kg KG/Tag reduziert werden. Bei unauffälligen Koronararterien in der Echokardiografie und normalisierter BSG kann ASS nach sechs bis acht Wochen sistiert werden. Die Therapie sollte möglichst innerhalb von sieben Tagen begonnen werden. Nach den aktuellen Leitlinien der deutschen Arbeitsgemeinschaft der wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) kann beim Vorhandensein von Fieber und mindestens vier der klinischen Kriterien am vierten Tag des Fiebers mit der Therapie begonnen werden. Das Risiko von Koronararterienveränderungen scheint bei einem früheren Therapiebeginn nicht weiter zu sinken; möglicherweise wird jedoch eine zweite IVIGGabe notwendig (5). Auch bei einer verzögerten Diagnosestellung, wie in unserem Fallbericht, scheint ein therapeutischer Nutzen gegeben. Etwa 10 bis 20 Prozent der Patienten sprechen auf eine einmalige Dosis IVIG und hochdosierte ASS nicht an. Diese Patienten fiebern 24 bis 36 Stunden nach der initialen IVIG-Gabe weiter und tragen ein besonders hohes Risiko für koronare Komplikationen (2). Das CRP bleibt weiter erhöht, das Albumin erniedrigt.
6 5/15
SCHWERPUNKT
Die Blutsenkungsreaktion kann in dieser Phase nicht als Parameter herangezogen werden, da diese durch die IVIG-Gabe erhöht sein kann (6). Bislang existieren keine randomisierten Studien zur Behandlung dieser Patienten; häufig wird eine zweite IVIG-Dosis (2 g/kg KG) gegeben. Etwa zwei Drittel der Patienten sprechen auf diese zweite IVIG-Gabe an. Bei weiter ausbleibendem Therapieeffekt spricht man von refraktärem oder IVIG-resistentem Kawasaki-Syndrom. In dieser Phase verbleiben verschiedene Optionen, darunter eine dritte IVIG-Gabe (2 g/kg KG), PulsSteroidgabe (20–30 mg/kg KG/Tag für 3 Tage) oder der TNF-alpha-Antagonist Infliximab (5 mg/kg KG); diese Massnahmen waren in Einzelstudien mit einem Therapieansprechen assoziiert. Einige Arbeiten der letzten Jahre weisen darauf hin, dass das Risiko für ein Nicht-Ansprechen auf die erste IVIG-Therapie bei bestimmten Patienten erhöht sein kann. Risikopatienten für das Versagen der einmaligen IVIG-Gabe weisen erniedrigte Serumnatriumwerte (≤ 133 mmol/l), erhöhte ASAT-Werte (> 100 U/l), eine Neutrophilie > 80 Prozent, CRP > 100 mg/l, Alter < 1 Jahr, initialer Therapiebeginn ≤ 4 Tage und Thrombozyten < 300 x 109/l auf. In einer randomisierten japanischen Studie wiesen Kobayashi et al. (7) nach, dass bei Vorliegen dieser Risikofaktoren nach einem vorgegebenen Punktschlüssel eine initiale Gabe von IVIG und gleichzeitiger intravenöser Prednisongabe (beginnend mit 2 mg/kg KG/Tag) das Auftreten einer Koronararterienbeteiligung senkte. Langzeitüberwachung Die Dauer und Art der Langzeitüberwachung richtet sich vorwiegend nach der Organbeteiligung in der Akutphase, vor allem den Koronaraneurysmen. Dabei werden nicht durchgehend einheitliche Standards verwendet; in den Empfehlungen von Newburger et al. (2) schlägt man eine Risikostratifizierung je nach Ausmass der Koronararterienbeteiligung vor. Ohne Koronaraneurysmen oder bei vorübergehenden Ektasien kann die ASS-Therapie nach sechs bis acht Wochen sistiert werden, Sport ist wieder ohne Ein- schränkungen möglich, eine Angiografie ist nicht indiziert, Kontrollen mit EKG und Echokardiografie werden nach 6, 12 und 60 Monaten wiederholt. Nach Aneurysmabildung stehen eine ASS-Langzeittherapie, engmaschigere EKG- und Echokontrollen, Belastungstests, Evaluation der sportlichen Aktivität und gegebenenfalls eine weitere Bildgebung (Angiografie, CT, MRI) im Vordergrund. Die Prognose wird hiervon massgeblich beeinflusst; die 10-Jahres-Überlebensrate liegt beim Vorhandensein grosser Aneurysmen bei etwa 95 Prozent (8). Das Kawasaki-Syndrom erfordert im Interesse des Patienten eine interdisziplinäre Vorgehensweise, um Komplikationsrisiken während der akuten Phase zu minimieren und die Langzeitprognose zu optimieren. Korrespondenzadresse: Elea Galiart Assistenzärztin Universitätskinderspital beider Basel (UKBB) Spitalstrasse 33 4031 Basel E-Mail: elea.galiart@ukbb.ch Literatur: 1. Ozen S et al.: EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis 2006; 65: 936–941. 2. Newburger JW et al.: Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics 2004; 114: 1708–1733. 3. Benseler SM et al.: Infections and Kawasaki disease: implications for coronary artery outcome. Pediatrics 2005; 116: e760–766. 4. Rigante D et al.: Kawasaki syndrome and concurrent Coxsackie virus B3 infection. Rheumatol Int 2012; 32: 4037–4040. 5. Muta H et al.: Early intravenous gamma-globulin treatment for Kawasaki disease: the nationwide surveys in Japan. J Pediatr 2004; 144: 496–499. 6. Hwang JY et al.: Assessment of intravenous immunoglobulin non-responders in Kawasaki disease. Arch Dis Child 2011; 96: 1088–1090. 7. Kobayashi T et al.: Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, openlabel, blinded-endpoints trial. Lancet 2012; 379: 1613–1620. 8. Kitamura S et al.: Twenty-five-year outcome of pediatric coronary artery bypass surgery for Kawasaki disease. Circulation 2009; 120: 60–68. 5/15