




Transkript
SCHWERPUNKT
Epileptische Enzephalopathien
Klinik und Genetik eines heterogenen Krankheitsspektrums
Epileptische Enzephalopathien sind ein heterogenes Krankheitsspektrum, das sich durch das Auftreten neurologischer beziehungsweise kognitiver Symptome in Verbindung mit epileptischer Aktivität auszeichnet. Die Diagnose des individuellen Syndroms ist wichtig für die Behandlung und Prognose. Durch den Wissenszuwachs der letzten Jahre sowie moderne Analysetechniken haben sich die Möglichkeiten zur Identifikation zugrunde liegender genetischer Defekte deutlich erhöht. Zudem zeigte sich eine klinische und genetische Überlappung der einzelnen Entitäten.
Von Johannes R. Lemke1, Sarah E. Bürki2, Alexandre N. Datta3
Die Diagnose des individuellen Syndroms ist wichtig für Behandlung und Prognose.
1Institut für Humangenetik, Universitätsklinikum Leipzig, Deutschland 2Pediatric Neurology, University of BC, BC Children's Hospital, Vancouver, British Columbia, Canada 3Abteilung für Neuro- und Entwicklungspädiatrie, Universitätskinderspital beider Basel (UKBB), Schweiz
20
B ei den epileptischen Enzephalopathien (EE) handelt es sich um ein Erkrankungsspektrum, bei dem vermutlich die epileptische Aktivität zur Ausprägung einer schweren kognitiven Beeinträchtigung sowie Verhaltensstörungen führt – und dies noch über das Mass hinaus, welches aufgrund einer zugrunde liegenden Pathologie (z.B. kortikale Malformationen) zu erwarten wäre (1). Die neuropsychologische Symptomatik ist typischerweise progredient. Bereits 2013 wurde eine Übersicht zur Einteilung neonataler und frühinfantiler EE erstellt (2). Die vorliegende Arbeit ergänzt diese Übersicht um spätinfantile Formen und komplettiert die zahlreichen neuen genetischen Erkenntnisse der jüngsten Vergangenheit.
Einteilung nach Erkrankungsalter
Die International League Against Epilepsy (ILAE) empfiehlt, Epilepsiesyndrome generell nach Erkrankungsalter einzuteilen (1). Die von der ILAE aufgeführten phänotypischen Merkmale einer epileptischen Enzephalopathie lassen klinisch und insbesondere auch hinsichtlich des Zeitpunkts der Erstmanifestation einen gewissen Spielraum für Entitäten zu, welche diese Kriterien erfüllen. In der vorliegenden Übersichtsarbeit möchten wir uns auf epileptische Enzephalopathien des Kindesalters beschränken, die durch die Abwesenheit nachweisbarer metabolischer Störungen und struktureller beziehungsweise läsioneller Hirnanomalien gekennzeichnet sind: Neugeborenenalter: • frühkindliche epileptische Enzephalopathie, Ohta-
hara-Syndrom (OS) • frühe myoklonische Enzephalopathie (FME) Säuglingsalter: • Epilepsie der frühen Kindheit mit migratorischen fo-
kalen Anfällen
• West-Syndrom (WS) • Dravet-Syndrom (DS) • myoklonische Enzephalopathie bei nicht progressi-
ven Störungen Kleinkind- und Schulalter: • Lennox-Gastaut-Syndrom (LGS) • Epilepsie mit myoklonisch-atonischen (früher asta-
tischen) Anfällen (Doose-Syndrom) • epileptische Enzephalopathie mit kontinuierlichen
Spike-Wave-Entladungen im Slow-Wave-Schlaf (CSWS / ESES) • Landau-Kleffner-Syndrom (LKS)
Frühkindliche epileptische Enzephalopathie
Der Begriff «Early Infantile Epileptic Encephalopathy» (EIEE) wurde ursprünglich von Ohtahara et al. 1976 etabliert und bezog sich auf ein frühkindliches Epilepsiesyndrom mit typischem EEG-Befund und Entwicklungsproblematik (3). Aufgrund allelischer Phänotypen führen nicht alle in Tabelle 1 gelisteten Mutationen zu einer EIEE; beispielsweise ist ein kleiner Anteil der dort erwähnten 1017 SCN1A-Mutationen für GEFS+, familiäre hemiplegische Migräne et cetera und nicht für das Dravet-Syndrom ursächlich, während ein Grossteil der bislang beschriebenen 126 KCNQ2- und der 66 SCN2A-Mutationen «benigne» Epilepsieformen verursacht. Frühkindliche epileptische Enzephalopathien (EIEE) werden in derzeit 18 Gruppen unterteilt, wobei diese Anzahl mit der Identifizierung neuer Gene kontinuierlich wächst (Tabelle 1). Einige Entitäten wurden dabei bislang nur in Einzelfällen beschrieben, andere belegen eine genetische Überlappung verschiedener Phänotypen beziehungsweise ihren Übergang ineinander.
3/14
SCHWERPUNKT
Ohtahara-Syndrom
Klinik: Dieses epileptische Syndrom wurde in der Erstbeschreibung von 1976 auch als frühinfantile epileptische Enzephalopathie bezeichnet (3). Es betrifft Neugeborene im Alter bis zu 3 Monaten und manifestiert sich mit häufigen tonischen Spasmen, aber auch anderen motorisch fokalen Anfällen (4). Das EEG zeigt ein charakteristisches Suppression-Burst-Muster. Der Verlauf ist schwer, mit erheblicher psychomotorischer Retardierung und therapieresistenten Anfällen. Der Begriff EIEE wird heute oft für ein breiteres Spektrum verwendet und beinhaltet nicht nur das OhtaharaSyndrom, sondern auch weitere ähnliche beziehungsweise überlappende Phänotypen (5). Etwa 75% der EIEE- oder Ohtahara-Patienten entwickeln im Verlauf ein West-Syndrom (6, 7). Mit bildgebenden Verfahren werden bei einem grossen Teil der Patienten grobe strukturelle Anomalien des Gehirns gefunden, wie zum Beispiel Hemimegalenzephalie, Agenesie des Corpus callosum, Porenzephalie und andere (8). Genetik: Systematische Analysen zum Mutationsspektrum von Ohtahara-Patienten liegen kaum vor. Die bislang bekannten genetischen Ursachen dieses Syndroms sind vielfältig und begründen sich zumeist auf nur wenige Fallbeschreibungen (Tabelle 2).
Neugeborenen- oder frühe myoklonische Enzephalopathie (EME)
Klinik: Zusammen mit der EIEE wird die Neugeborenen- oder frühe Myoklonus-Enzephalopathie (engl. Synonym: EME) in die Gruppe der epileptischen Enzephalopathien mit Suppression-Burst-EEG eingeteilt. Die EME wurde erstmals 1978 durch J. Aicardi und F. Goutières (16) zwei Jahre nach dem Othahara-Syndrom, beschrieben. Die Anfälle beginnen ebenfalls sehr früh, innerhalb der ersten 3 Lebensmonate, zum Teil schon wenige Stunden nach Geburt. Typischerweise handelt es sich um fokale Myoklonien der Extremitäten oder aber auch des Gesichts/der Augenlider ohne EEG-Korrelat. Sie werden oft als sogenannt erratisch oder fragmentiert beschrieben. Daneben haben über 80 Prozent der Kinder auch fokale Anfälle, die sehr subtil sein können und sich beispielsweise nur durch kurze Augendeviationen, diskrete autonome Zeichen (Flush des Gesichts) oder Apnoen manifestieren. Das Suppression-Burst-Muster im EEG (Abbildung 1) kann sich erst nach Anfallsbeginn manifestieren und ist im Unterschied zum Othahara-Syndrom nicht kontinuierlich und im Schlaf akzentuiert. Im Alter von 3 bis 5 Monaten kann sich das elektroenzephalographische Bild bei bis 50 Prozent der Patienten in eine atypische Hypsarrhythmie ändern (17). Bis zur Hälfte der Kinder versterben vor dem 2. Lebensjahr. Die anderen zeigen eine schwere globale Entwicklungsstörung und bleiben zum Teil in einem vegetativen Zustand (18). Wie bei der EIEE ist die Pathogenese der EME sehr variabel, und sowohl strukturelle, metabolische und auch genetische Veränderungen können eine Rolle spielen. Meist scheint es sich jedoch um einen diffusen Prozess zu handeln, der vor allem den Hirnstamm und die weisse Substanz involviert. Dies führt möglicherweise zu einer Deafferenzierung und Hyperexzitabilität des Cortex, was
wiederum am ehesten suggestiv für eine zugrunde liegende metabolische oder degenerative Erkrankung ist (19). Dazu passend werden als häufige Ätiologie eine nichtketotische Hyperglycinämie, aber auch andere metabolische Störungen wie Proprionacidurie oder Molybdän-Kofaktormangel beschrieben (8).
Tabelle 1:
Phänotypen frühkindlicher epileptischer Enzephalopathien (EIEE)
OMIM- Gen Entität
Anzahl beschriebener Mutationen
Allelische Phänotypen (neben EIEE)
EIEE1 ARX
64
EIEE2 EIEE3 EIEE4 EIEE5 EIEE6
CDKL5 201 SLC25A22 2 STXBP1 68 SPTAN1 8 SCN1A 1017
EIEE7 EIEE8 EIEE9 EIEE10 EIEE11 EIEE12 EIEE13 EIEE14 EIEE15 EIEE16
KCNQ2 ARHGEF9 PCDH19 PNKP SCN2A PLCB1 SCN8A KCNT1 ST3GAL3 TBC1D24
126 8 114 5 66 5 5 11 3 6
EIEE17 EIEE18
GNAO1 SZT2
4 3
Hydranencephaly with abnormal genitalia, X-linked lissencephaly 2, X-linked mental retardation 29, Partington syndrome, Proud syndrome Rett syndrome-like, Angelman syndrome-like
Dravet syndrome, generalized epilepsy with febrile seizures plus type 2, familial febrile seizures 3A, familial hemiplegic migraine 3 benign neonatal seizures 1, Myokymia
benign familial infantile seizures 3
cognitive impairment with or without cerebellar ataxia nocturnal frontal lobe epilepsy 3 autosomal recessive mental retardation 12 DOOR syndrome, autosomal recessive deafness 86, familial infantile myoclonic epilepsy
Die Tabelle umfasst derzeit bekannte EIEE-Phänotypen gemäss OMIM-Datenbank (Online Mendelian Inheritance in Man) mit ihren jeweils ursächlichen Genen sowie die Anzahl der für diese Gene derzeit beschriebenen Mutationen laut Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/index.php); Stand: März 2014
Tabelle 2:
Mutationen bei Ohtahara-Syndrom
Mutation
Phänotyp
ARX STXBP1
KCNQ2
copy number variations
männliche Ohtahara-Patienten mit zusätzlichen Befunden wie Genitalanomalien, Corpus-callosum-Agenesie, Lissencephalie (9), progressiver Mikrozephalie (10) und/oder choreatischer Dystonie (11) keine signifikanten Fehlbildungen und/oder Dysmorphien, häufig frühzeitige Bewegungsstörung, oft deutliche Besserung der Anfallssituation in ersten Lebensjahren, Mutationsnachweis bei 2 von 6 (33%) Ohtahara-Patienten bzw. 12 von 266 (4,5%) EIEE-Patienten (12, 13) keine signifikanten Fehlbildungen und/oder Dysmorphien, oft deutliche Besserung der Anfallssituation in den ersten Lebensjahren, Mutationsnachweis bei 19 von 164 (11,6%) EIEE-Patienten (14, 15) Die Mehrheit der KCNQ2-Mutationsträger entwickelt jedoch BFNE und keine epileptische Enzephalopathie. variabler Phänotyp, zumeist individuelle Mikrodeletion
3/14
21
SCHWERPUNKT
Genetik: Die genetischen Ursachen der EME sind unklar. In der Literatur findet sich ein Einzelfallbericht eines EME-Patienten mit einer de novo reziproken chromosomalen Translokation und konsekutiver Dysruption des ERBB4-Gens, welches möglicherweise eine Rolle bei der Neuromigration spielt (20).
Epilepsie der frühen Kindheit mit
migratorischen fokalen Anfällen
(MMPEI)
Klinik: Seit der Erstbeschreibung des Syndroms durch
Coppola et al. 1995 (21) wurden etwa einige Dutzend
Fälle publiziert (22). 2001 wurde die MMPEI (Mali-
gnant Migrating Partial Epilepsy in Infancy) erstmals
in der ILAE erwähnt (23). Der natürliche klinische Ver-
lauf zeigt typischerweise 3 Phasen:
1. Phase: Während der ersten
3 Lebensmonate treten ver-
einzelte, vor allem fokale An-
fälle mit schneller sekundärer
Generalisierung auf, häufig
begleitet von autonomen
Symptomen (Apnoe, Flush,
Zyanose). Die Dauer dieser
Phase beträgt wenige Wo-
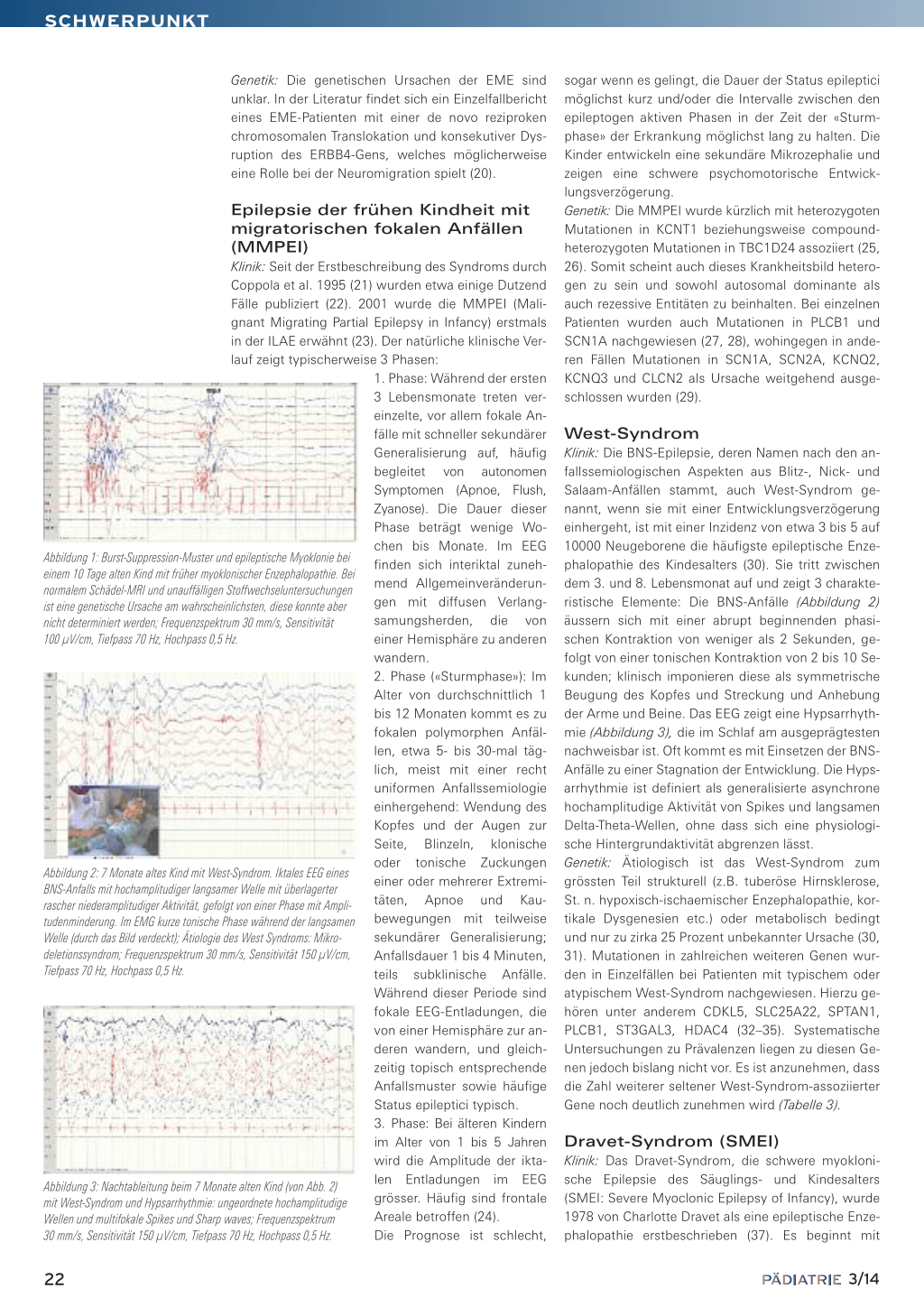
Abbildung 1: Burst-Suppression-Muster und epileptische Myoklonie bei einem 10 Tage alten Kind mit früher myoklonischer Enzephalopathie. Bei normalem Schädel-MRI und unauffälligen Stoffwechseluntersuchungen ist eine genetische Ursache am wahrscheinlichsten, diese konnte aber nicht determiniert werden; Frequenzspektrum 30 mm/s, Sensitivität 100 µV/cm, Tiefpass 70 Hz, Hochpass 0,5 Hz.
chen bis Monate. Im EEG finden sich interiktal zunehmend Allgemeinveränderungen mit diffusen Verlangsamungsherden, die von einer Hemisphäre zu anderen
wandern.
2. Phase («Sturmphase»): Im
Alter von durchschnittlich 1
bis 12 Monaten kommt es zu
fokalen polymorphen Anfäl-
len, etwa 5- bis 30-mal täg-
lich, meist mit einer recht
uniformen Anfallssemiologie
einhergehend: Wendung des
Kopfes und der Augen zur
Seite, Blinzeln, klonische
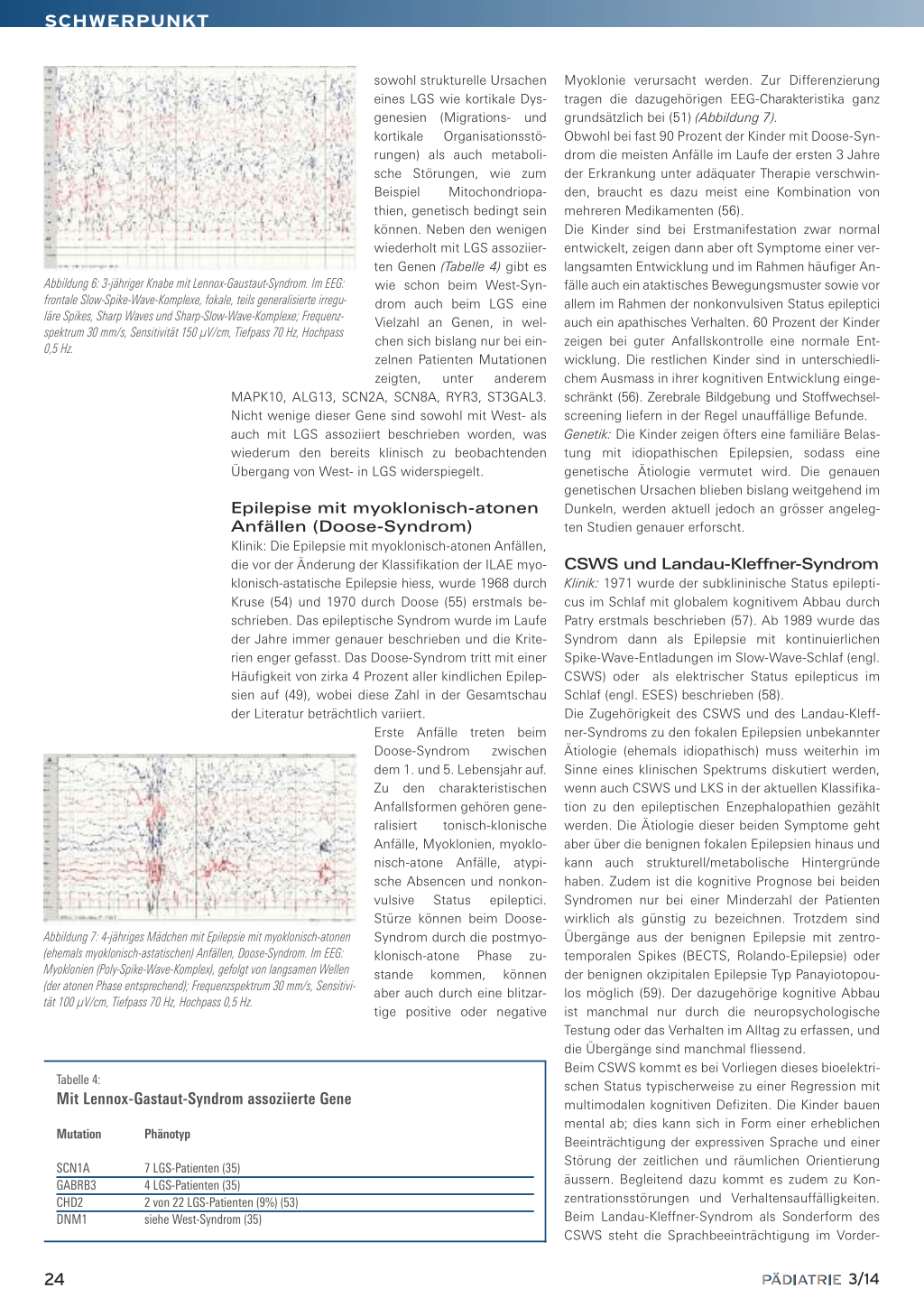
Abbildung 2: 7 Monate altes Kind mit West-Syndrom. Iktales EEG eines BNS-Anfalls mit hochamplitudiger langsamer Welle mit überlagerter rascher niederamplitudiger Aktivität, gefolgt von einer Phase mit Amplitudenminderung. Im EMG kurze tonische Phase während der langsamen Welle (durch das Bild verdeckt); Ätiologie des West Syndroms: Mikro-
oder tonische Zuckungen einer oder mehrerer Extremitäten, Apnoe und Kaubewegungen mit teilweise sekundärer Generalisierung;
deletionssyndrom; Frequenzspektrum 30 mm/s, Sensitivität 150 µV/cm, Anfallsdauer 1 bis 4 Minuten,
Tiefpass 70 Hz, Hochpass 0,5 Hz.
teils subklinische Anfälle.
Während dieser Periode sind
fokale EEG-Entladungen, die
von einer Hemisphäre zur an-
deren wandern, und gleich-
zeitig topisch entsprechende
Anfallsmuster sowie häufige
Status epileptici typisch.
3. Phase: Bei älteren Kindern
im Alter von 1 bis 5 Jahren
wird die Amplitude der ikta-
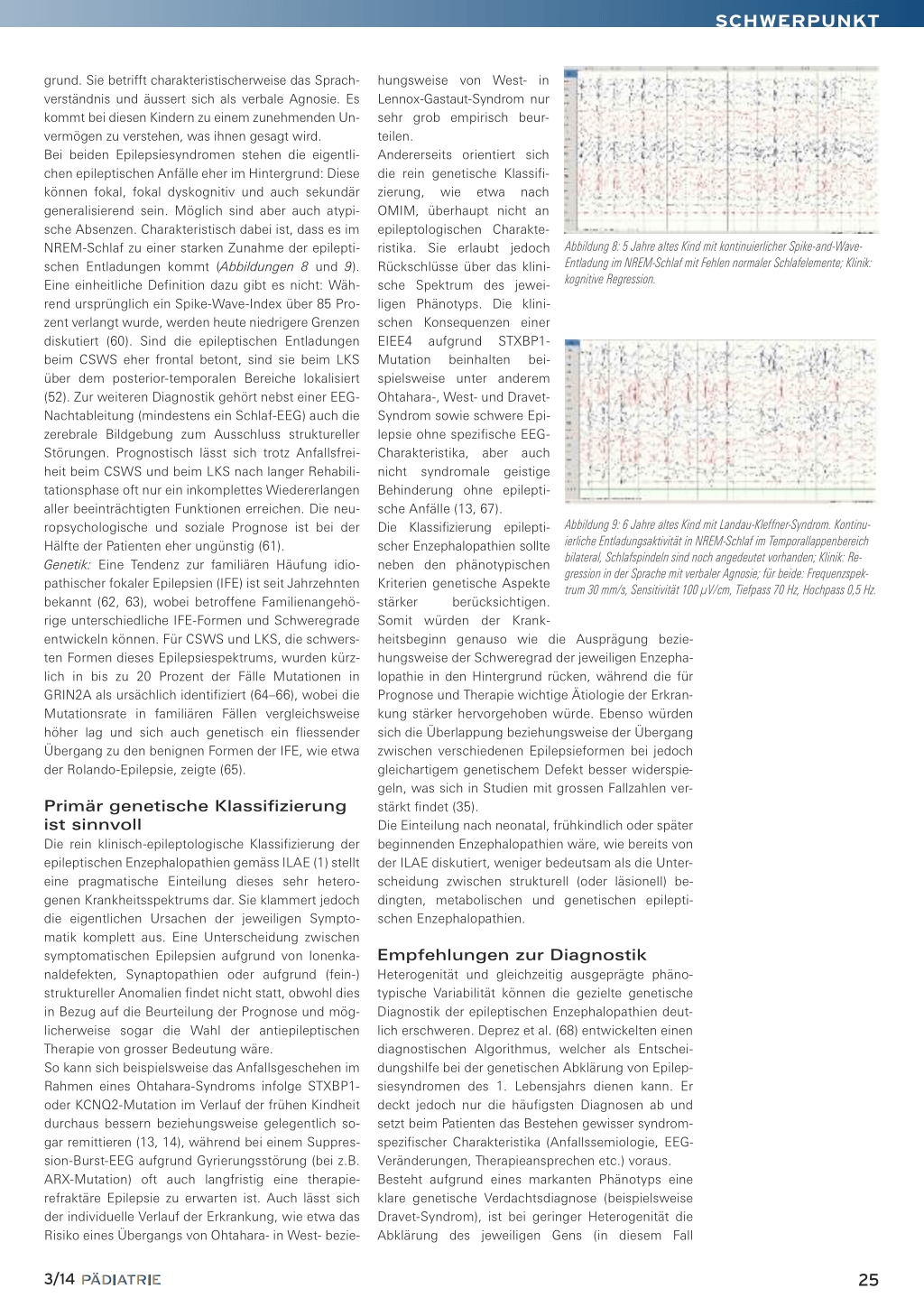
Abbildung 3: Nachtableitung beim 7 Monate alten Kind (von Abb. 2) mit West-Syndrom und Hypsarrhythmie: ungeordnete hochamplitudige Wellen und multifokale Spikes und Sharp waves; Frequenzspektrum 30 mm/s, Sensitivität 150 µV/cm, Tiefpass 70 Hz, Hochpass 0,5 Hz.
len Entladungen im EEG grösser. Häufig sind frontale Areale betroffen (24). Die Prognose ist schlecht,
sogar wenn es gelingt, die Dauer der Status epileptici möglichst kurz und/oder die Intervalle zwischen den epileptogen aktiven Phasen in der Zeit der «Sturmphase» der Erkrankung möglichst lang zu halten. Die Kinder entwickeln eine sekundäre Mikrozephalie und zeigen eine schwere psychomotorische Entwicklungsverzögerung. Genetik: Die MMPEI wurde kürzlich mit heterozygoten Mutationen in KCNT1 beziehungsweise compoundheterozygoten Mutationen in TBC1D24 assoziiert (25, 26). Somit scheint auch dieses Krankheitsbild heterogen zu sein und sowohl autosomal dominante als auch rezessive Entitäten zu beinhalten. Bei einzelnen Patienten wurden auch Mutationen in PLCB1 und SCN1A nachgewiesen (27, 28), wohingegen in anderen Fällen Mutationen in SCN1A, SCN2A, KCNQ2, KCNQ3 und CLCN2 als Ursache weitgehend ausgeschlossen wurden (29).
West-Syndrom
Klinik: Die BNS-Epilepsie, deren Namen nach den anfallssemiologischen Aspekten aus Blitz-, Nick- und Salaam-Anfällen stammt, auch West-Syndrom genannt, wenn sie mit einer Entwicklungsverzögerung einhergeht, ist mit einer Inzidenz von etwa 3 bis 5 auf 10000 Neugeborene die häufigste epileptische Enzephalopathie des Kindesalters (30). Sie tritt zwischen dem 3. und 8. Lebensmonat auf und zeigt 3 charakteristische Elemente: Die BNS-Anfälle (Abbildung 2) äussern sich mit einer abrupt beginnenden phasischen Kontraktion von weniger als 2 Sekunden, gefolgt von einer tonischen Kontraktion von 2 bis 10 Sekunden; klinisch imponieren diese als symmetrische Beugung des Kopfes und Streckung und Anhebung der Arme und Beine. Das EEG zeigt eine Hypsarrhythmie (Abbildung 3), die im Schlaf am ausgeprägtesten nachweisbar ist. Oft kommt es mit Einsetzen der BNSAnfälle zu einer Stagnation der Entwicklung. Die Hypsarrhythmie ist definiert als generalisierte asynchrone hochamplitudige Aktivität von Spikes und langsamen Delta-Theta-Wellen, ohne dass sich eine physiologische Hintergrundaktivität abgrenzen lässt. Genetik: Ätiologisch ist das West-Syndrom zum grössten Teil strukturell (z.B. tuberöse Hirnsklerose, St. n. hypoxisch-ischaemischer Enzephalopathie, kortikale Dysgenesien etc.) oder metabolisch bedingt und nur zu zirka 25 Prozent unbekannter Ursache (30, 31). Mutationen in zahlreichen weiteren Genen wurden in Einzelfällen bei Patienten mit typischem oder atypischem West-Syndrom nachgewiesen. Hierzu gehören unter anderem CDKL5, SLC25A22, SPTAN1, PLCB1, ST3GAL3, HDAC4 (32–35). Systematische Untersuchungen zu Prävalenzen liegen zu diesen Genen jedoch bislang nicht vor. Es ist anzunehmen, dass die Zahl weiterer seltener West-Syndrom-assoziierter Gene noch deutlich zunehmen wird (Tabelle 3).
Dravet-Syndrom (SMEI)
Klinik: Das Dravet-Syndrom, die schwere myoklonische Epilepsie des Säuglings- und Kindesalters (SMEI: Severe Myoclonic Epilepsy of Infancy), wurde 1978 von Charlotte Dravet als eine epileptische Enzephalopathie erstbeschrieben (37). Es beginnt mit
22 3/14
SCHWERPUNKT
Anfällen im Alter von 3 bis 12 Monaten bei vorgängig gesunden, altersentsprechend entwickelten Kindern. Meistens präsentieren sie sich zu Beginn mit einem febrilen Status epilepticus und/oder hemiklonischen Anfällen mit und ohne Fieber. Im Alter von 1 bis 4 Jahren kommen vor allem fokal dyskognitive Anfälle, kurze (< 10 Sek.) atypische Absenzen, zum Teil in Clustern, und bei einem Teil der Kinder myoklonische Anfälle hinzu (Abbildung 4). Trigger sind klassischerweise Fieber, Müdigkeit und Photostimulation bei photosensiblen Patienten. Das EEG ist zu Beginn häufig normal und ab etwa dem 2. Lebensjahr oft pathologisch mit einer generalisierten Verlangsamung (Abbildung 5) und multifokalen epileptiformen Veränderungen. Neuroradiologisch lassen sich, wenn überhaupt, nur unspezifische Auffälligkeiten (z.B. Atrophie) und bei etwa 30 Prozent der Kinder eine Hippocampussklerose nachweisen. Ab dem 2. Lebensjahr zeigen sich in der Regel motorische und kognitive Defizite sowie Verhaltensauffälligkeiten. Die Kinder haben oft einen späten Gehbeginn (durchschnittlich mit 17 Monaten) und eine Ataxie. Tendenziell treten ab zirka dem 9. Lebensjahr zunehmend Gehschwierigkeiten (sog. «crouch pattern») auf (38). Die Wahl einer adäquaten bzw. spezifischen antiepileptischen Therapie hat einen Einfluss auf die individuelle Prognose eines Dravet-Patienten (39). Genetik: Die genetische Ursache des Dravet-Syndroms liegt in mindestens 75 Prozent der Fälle in Mutationen der Natriumkanaluntereinheit SCN1A (40). Bei Patienten mit Dravet-ähnlichen Phänotypen finden sich gelegentlich Mutationen in weiteren Genen, wie etwa PCDH19 (bei Mädchen) (41) oder seltener SCN1B, SCN2A, GABRG2. Kürzlich konnten bei einigen Dravet-Patienten auch Mutationen in CHD2, GABRA1 und STXBP1 nachgewiesen werden (42, 43). Somit besteht bei SCN1A-negativen Dravet-Patienten eine relativ hohe genetische Heterogenität. Mutationen in SCN9A werden als modifizierende Faktoren des Dravet-Syndroms gewertet und wurden in mehreren Fällen zusätzlich zu einer SCN1A-Mutation nachgewiesen (44). Bei Patienten mit typischem Dravet-Syndrom ohne nachweisbare SCN1A-Mutation kann es sich lohnen, den Originalbefund zu hinterfragen, da neueste Sequenziertechniken wiederholt falsch-negative Befunde in der klassischen Routinediagnostik aufdeckten (45). Zumeist waren diese auf ältere beziehungsweise weniger sensitive Analysemethoden in der primären Abklärung zurückzuführen, aber selten auch auf methodische Artefakte der Sanger-Sequenzierung. postnataler Schädigung, ze- rebralen Malformationen, progressiven Enzephalopa- thien oder auch chromoso- malen Störungen zuzuord- nen. Übergänge aus einem anderen Epilepsiesyndrom wie Ohtahara- oder West- Syndrom sind dabei nicht selten. Klinisch manifestiert sich das LGS charakteristischerweise durch eine Vielzahl verschie- Abbildung 4: 6-jähriger Knabe mit Dravet-Syndrom. Im EEG: 2 Myoklonien (Pfeile) als Poly-Spike-Wave-Komplexe sichtbar, gefolgt von einer kurzen atypischen Absenz mit langsamen Wellen; für beide: Frequenzspektrum 30 mm/s, Sensitivität 100 µV/cm, Tiefpass 70 Hz, Hochpass 0,5 Hz. denener Anfallsformen: Toni- sche Anfälle mit oder ohne vegetative Begleitsymptome (Gesichtsrötung, Tachykar- die, Pupillendilatation) stellen die Hauptanfallsform dar und treten tags, aber typischer- weise auch nachts auf, da der Tiefschlaf deren Auftreten fördert. Die kurzen axialen to- nischen Anfälle tagsüber füh- ren oft auch zu Stürzen. Stürze sind beim LGS auch im Rahmen der atonischen Abbildung 5: 6-jähriger Knabe mit Dravet-Syndrom. Im EEG: etwas langsame Theta-Aktivität im Zentralbereich, Beta-Wellen-Überlagerung (durch Benzodiazepine). Anfälle möglich, wobei diese teils ebenfalls mit Myoklo- nien assoziiert sind. Atypische Absenzen von längerer Dauer und nicht konvulsive Status epileptici sind weitere Anfallsformen des LGS (50). Das klassische EEG-Merkmal des LGS (Abbildung 6) sind bilaterale frontale 1,5- bis 2-Slow-Spike-Wave-Komplexe im Wachzustand sowie bilaterale synchrone rasche Rhythmen im NREM-Schlaf, teils einhergehend mit kurzen tonischen Anfallssymptomen (51, 52). Die Therapie bei Lennox-Gastaut-Syndrom stellt eine äusserst grosse Herausforderung dar. Sie ist aufgrund mangelnden Erfolgs meist nicht zufriedenstellend. Prognostisch bleibt das Lennox-Gastaut-Syndrom un- günstig: Nur 7 bis 15 Prozent aller Patienten mit LGS entwickeln sich normal oder sind mental leicht behin- dert, die anderen sind in der Regel schwer betroffen. Genetik: Die Ätiologie des LGS umfasst die ganze Bandbreite struktureller, metabolischer und primär genetischer Pathologien. Dabei ist zu beachten, dass Tabelle 3: Mutationen bei West-Syndrom Lennox-Gastaut-Syndrom (LGS) Klinik: Das Lennox-Gastaut-Syndrom wurde 1950 von Lennox und Davis (46) und 1966 von Gastaut (47) beschrieben und gehört zu den am schwersten verlaufenden epileptischen Enzephalopathien des Kindesalters. Es macht allerdings nur 1 bis 2 Prozent aller Epilepsien im Kindes- und Jugendalter aus (48, 49). Das Syndrom manifestiert sich meist im Alter von 3 bis 6 Jahren (mögliche Altersspanne 1 bis 10 Jahre) und ist in drei Viertel der Fälle einer strukturellen/metabolischen oder genetischen Ätiologie, peri- oder Mutation ARX STXBP1 DNM1 GRIN2B copy number variations Phänotyp siehe Ohtahara-Syndrom keine signifikanten Fehlbildungen und/oder Dysmorphien, Mutationsnachweis bei 1 von 65 (2%) West-Patienten (13), 4 von 6 (67%) STXBP1-Mutationsträgern entwickelten ein West-Syndrom (12) West-Syndrom mit Übergang in Lennox-Gastaut-Syndrom sowie weitere nichtläsionelle EE-Formen (35) West-Syndrom mit schwerer geistiger Behinderung (36) variabler Phänotyp, zumeist individuelle Mikrodeletion 3/14 23 SCHWERPUNKT sowohl strukturelle Ursachen eines LGS wie kortikale Dys- genesien (Migrations- und kortikale Organisationsstö- rungen) als auch metaboli- sche Störungen, wie zum Beispiel Mitochondriopa- thien, genetisch bedingt sein können. Neben den wenigen wiederholt mit LGS assoziier- Abbildung 6: 3-jähriger Knabe mit Lennox-Gaustaut-Syndrom. Im EEG: frontale Slow-Spike-Wave-Komplexe, fokale, teils generalisierte irreguläre Spikes, Sharp Waves und Sharp-Slow-Wave-Komplexe; Frequenzspektrum 30 mm/s, Sensitivität 150 µV/cm, Tiefpass 70 Hz, Hochpass 0,5 Hz. ten Genen (Tabelle 4) gibt es wie schon beim West-Syndrom auch beim LGS eine Vielzahl an Genen, in welchen sich bislang nur bei einzelnen Patienten Mutationen zeigten, unter anderem MAPK10, ALG13, SCN2A, SCN8A, RYR3, ST3GAL3. Nicht wenige dieser Gene sind sowohl mit West- als auch mit LGS assoziiert beschrieben worden, was wiederum den bereits klinisch zu beobachtenden Übergang von West- in LGS widerspiegelt. Epilepise mit myoklonisch-atonen Anfällen (Doose-Syndrom) Klinik: Die Epilepsie mit myoklonisch-atonen Anfällen, die vor der Änderung der Klassifikation der ILAE myo- klonisch-astatische Epilepsie hiess, wurde 1968 durch Kruse (54) und 1970 durch Doose (55) erstmals be- schrieben. Das epileptische Syndrom wurde im Laufe der Jahre immer genauer beschrieben und die Krite- rien enger gefasst. Das Doose-Syndrom tritt mit einer Häufigkeit von zirka 4 Prozent aller kindlichen Epilep- sien auf (49), wobei diese Zahl in der Gesamtschau der Literatur beträchtlich variiert. Erste Anfälle treten beim Doose-Syndrom zwischen dem 1. und 5. Lebensjahr auf. Zu den charakteristischen Anfallsformen gehören gene- ralisiert tonisch-klonische Anfälle, Myoklonien, myoklo- nisch-atone Anfälle, atypi- sche Absencen und nonkon- vulsive Status epileptici. Stürze können beim Doose- Abbildung 7: 4-jähriges Mädchen mit Epilepsie mit myoklonisch-atonen (ehemals myoklonisch-astatischen) Anfällen, Doose-Syndrom. Im EEG: Myoklonien (Poly-Spike-Wave-Komplex), gefolgt von langsamen Wellen (der atonen Phase entsprechend); Frequenzspektrum 30 mm/s, Sensitivität 100 µV/cm, Tiefpass 70 Hz, Hochpass 0,5 Hz. Syndrom durch die postmyoklonisch-atone Phase zustande kommen, können aber auch durch eine blitzartige positive oder negative Tabelle 4: Mit Lennox-Gastaut-Syndrom assoziierte Gene Mutation Phänotyp SCN1A GABRB3 CHD2 DNM1 7 LGS-Patienten (35) 4 LGS-Patienten (35) 2 von 22 LGS-Patienten (9%) (53) siehe West-Syndrom (35) Myoklonie verursacht werden. Zur Differenzierung tragen die dazugehörigen EEG-Charakteristika ganz grundsätzlich bei (51) (Abbildung 7). Obwohl bei fast 90 Prozent der Kinder mit Doose-Syndrom die meisten Anfälle im Laufe der ersten 3 Jahre der Erkrankung unter adäquater Therapie verschwinden, braucht es dazu meist eine Kombination von mehreren Medikamenten (56). Die Kinder sind bei Erstmanifestation zwar normal entwickelt, zeigen dann aber oft Symptome einer verlangsamten Entwicklung und im Rahmen häufiger Anfälle auch ein ataktisches Bewegungsmuster sowie vor allem im Rahmen der nonkonvulsiven Status epileptici auch ein apathisches Verhalten. 60 Prozent der Kinder zeigen bei guter Anfallskontrolle eine normale Entwicklung. Die restlichen Kinder sind in unterschiedlichem Ausmass in ihrer kognitiven Entwicklung eingeschränkt (56). Zerebrale Bildgebung und Stoffwechselscreening liefern in der Regel unauffällige Befunde. Genetik: Die Kinder zeigen öfters eine familiäre Belastung mit idiopathischen Epilepsien, sodass eine genetische Ätiologie vermutet wird. Die genauen genetischen Ursachen blieben bislang weitgehend im Dunkeln, werden aktuell jedoch an grösser angelegten Studien genauer erforscht. CSWS und Landau-Kleffner-Syndrom Klinik: 1971 wurde der subklininische Status epilepticus im Schlaf mit globalem kognitivem Abbau durch Patry erstmals beschrieben (57). Ab 1989 wurde das Syndrom dann als Epilepsie mit kontinuierlichen Spike-Wave-Entladungen im Slow-Wave-Schlaf (engl. CSWS) oder als elektrischer Status epilepticus im Schlaf (engl. ESES) beschrieben (58). Die Zugehörigkeit des CSWS und des Landau-Kleffner-Syndroms zu den fokalen Epilepsien unbekannter Ätiologie (ehemals idiopathisch) muss weiterhin im Sinne eines klinischen Spektrums diskutiert werden, wenn auch CSWS und LKS in der aktuellen Klassifikation zu den epileptischen Enzephalopathien gezählt werden. Die Ätiologie dieser beiden Symptome geht aber über die benignen fokalen Epilepsien hinaus und kann auch strukturell/metabolische Hintergründe haben. Zudem ist die kognitive Prognose bei beiden Syndromen nur bei einer Minderzahl der Patienten wirklich als günstig zu bezeichnen. Trotzdem sind Übergänge aus der benignen Epilepsie mit zentrotemporalen Spikes (BECTS, Rolando-Epilepsie) oder der benignen okzipitalen Epilepsie Typ Panayiotopoulos möglich (59). Der dazugehörige kognitive Abbau ist manchmal nur durch die neuropsychologische Testung oder das Verhalten im Alltag zu erfassen, und die Übergänge sind manchmal fliessend. Beim CSWS kommt es bei Vorliegen dieses bioelektrischen Status typischerweise zu einer Regression mit multimodalen kognitiven Defiziten. Die Kinder bauen mental ab; dies kann sich in Form einer erheblichen Beeinträchtigung der expressiven Sprache und einer Störung der zeitlichen und räumlichen Orientierung äussern. Begleitend dazu kommt es zudem zu Konzentrationsstörungen und Verhaltensauffälligkeiten. Beim Landau-Kleffner-Syndrom als Sonderform des CSWS steht die Sprachbeeinträchtigung im Vorder- 24 3/14 SCHWERPUNKT grund. Sie betrifft charakteristischerweise das Sprachverständnis und äussert sich als verbale Agnosie. Es kommt bei diesen Kindern zu einem zunehmenden Unvermögen zu verstehen, was ihnen gesagt wird. Bei beiden Epilepsiesyndromen stehen die eigentlichen epileptischen Anfälle eher im Hintergrund: Diese können fokal, fokal dyskognitiv und auch sekundär generalisierend sein. Möglich sind aber auch atypische Absenzen. Charakteristisch dabei ist, dass es im NREM-Schlaf zu einer starken Zunahme der epileptischen Entladungen kommt (Abbildungen 8 und 9). Eine einheitliche Definition dazu gibt es nicht: Während ursprünglich ein Spike-Wave-Index über 85 Prozent verlangt wurde, werden heute niedrigere Grenzen diskutiert (60). Sind die epileptischen Entladungen beim CSWS eher frontal betont, sind sie beim LKS über dem posterior-temporalen Bereiche lokalisiert (52). Zur weiteren Diagnostik gehört nebst einer EEGNachtableitung (mindestens ein Schlaf-EEG) auch die zerebrale Bildgebung zum Ausschluss struktureller Störungen. Prognostisch lässt sich trotz Anfallsfreiheit beim CSWS und beim LKS nach langer Rehabilitationsphase oft nur ein inkomplettes Wiedererlangen aller beeinträchtigten Funktionen erreichen. Die neuropsychologische und soziale Prognose ist bei der Hälfte der Patienten eher ungünstig (61). Genetik: Eine Tendenz zur familiären Häufung idiopathischer fokaler Epilepsien (IFE) ist seit Jahrzehnten bekannt (62, 63), wobei betroffene Familienangehörige unterschiedliche IFE-Formen und Schweregrade entwickeln können. Für CSWS und LKS, die schwersten Formen dieses Epilepsiespektrums, wurden kürzlich in bis zu 20 Prozent der Fälle Mutationen in GRIN2A als ursächlich identifiziert (64–66), wobei die Mutationsrate in familiären Fällen vergleichsweise höher lag und sich auch genetisch ein fliessender Übergang zu den benignen Formen der IFE, wie etwa der Rolando-Epilepsie, zeigte (65). Primär genetische Klassifizierung ist sinnvoll Die rein klinisch-epileptologische Klassifizierung der epileptischen Enzephalopathien gemäss ILAE (1) stellt eine pragmatische Einteilung dieses sehr heterogenen Krankheitsspektrums dar. Sie klammert jedoch die eigentlichen Ursachen der jeweiligen Symptomatik komplett aus. Eine Unterscheidung zwischen symptomatischen Epilepsien aufgrund von Ionenkanaldefekten, Synaptopathien oder aufgrund (fein-) struktureller Anomalien findet nicht statt, obwohl dies in Bezug auf die Beurteilung der Prognose und möglicherweise sogar die Wahl der antiepileptischen Therapie von grosser Bedeutung wäre. So kann sich beispielsweise das Anfallsgeschehen im Rahmen eines Ohtahara-Syndroms infolge STXBP1oder KCNQ2-Mutation im Verlauf der frühen Kindheit durchaus bessern beziehungsweise gelegentlich sogar remittieren (13, 14), während bei einem Suppression-Burst-EEG aufgrund Gyrierungsstörung (bei z.B. ARX-Mutation) oft auch langfristig eine therapierefraktäre Epilepsie zu erwarten ist. Auch lässt sich der individuelle Verlauf der Erkrankung, wie etwa das Risiko eines Übergangs von Ohtahara- in West- bezie- hungsweise von West- in Lennox-Gastaut-Syndrom nur sehr grob empirisch beur- teilen. Andererseits orientiert sich die rein genetische Klassifi- zierung, wie etwa nach OMIM, überhaupt nicht an epileptologischen Charakte- ristika. Sie erlaubt jedoch Rückschlüsse über das klinische Spektrum des jewei- Abbildung 8: 5 Jahre altes Kind mit kontinuierlicher Spike-and-WaveEntladung im NREM-Schlaf mit Fehlen normaler Schlafelemente; Klinik: kognitive Regression. ligen Phänotyps. Die klini- schen Konsequenzen einer EIEE4 aufgrund STXBP1- Mutation beinhalten bei- spielsweise unter anderem Ohtahara-, West- und Dravet- Syndrom sowie schwere Epi- lepsie ohne spezifische EEG- Charakteristika, aber auch nicht syndromale geistige Behinderung ohne epilepti- sche Anfälle (13, 67). Die Klassifizierung epilepti- scher Enzephalopathien sollte neben den phänotypischen Kriterien genetische Aspekte stärker berücksichtigen. Abbildung 9: 6 Jahre altes Kind mit Landau-Kleffner-Syndrom. Kontinuierliche Entladungsaktivität in NREM-Schlaf im Temporallappenbereich bilateral, Schlafspindeln sind noch angedeutet vorhanden; Klinik: Regression in der Sprache mit verbaler Agnosie; für beide: Frequenzspektrum 30 mm/s, Sensitivität 100 µV/cm, Tiefpass 70 Hz, Hochpass 0,5 Hz. Somit würden der Krank- heitsbeginn genauso wie die Ausprägung bezie- hungsweise der Schweregrad der jeweiligen Enzepha- lopathie in den Hintergrund rücken, während die für Prognose und Therapie wichtige Ätiologie der Erkran- kung stärker hervorgehoben würde. Ebenso würden sich die Überlappung beziehungsweise der Übergang zwischen verschiedenen Epilepsieformen bei jedoch gleichartigem genetischem Defekt besser widerspie- geln, was sich in Studien mit grossen Fallzahlen ver- stärkt findet (35). Die Einteilung nach neonatal, frühkindlich oder später beginnenden Enzephalopathien wäre, wie bereits von der ILAE diskutiert, weniger bedeutsam als die Unter- scheidung zwischen strukturell (oder läsionell) be- dingten, metabolischen und genetischen epilepti- schen Enzephalopathien. Empfehlungen zur Diagnostik Heterogenität und gleichzeitig ausgeprägte phänotypische Variabilität können die gezielte genetische Diagnostik der epileptischen Enzephalopathien deutlich erschweren. Deprez et al. (68) entwickelten einen diagnostischen Algorithmus, welcher als Entscheidungshilfe bei der genetischen Abklärung von Epilepsiesyndromen des 1. Lebensjahrs dienen kann. Er deckt jedoch nur die häufigsten Diagnosen ab und setzt beim Patienten das Bestehen gewisser syndromspezifischer Charakteristika (Anfallssemiologie, EEGVeränderungen, Therapieansprechen etc.) voraus. Besteht aufgrund eines markanten Phänotyps eine klare genetische Verdachtsdiagnose (beispielsweise Dravet-Syndrom), ist bei geringer Heterogenität die Abklärung des jeweiligen Gens (in diesem Fall 3/14 25 SCHWERPUNKT SCN1A) der wohl effizienteste Weg, die Diagnose genetisch zu sichern. Etwa 8 Prozent aller EE-Patienten haben nachweisbare submikroskopische Chromosomenaberrationen (69), weswegen eine Microarray-Analyse (Array-CGH oder SNP-Chip) immer indiziert ist. Mittels moderner Hochdurchsatzsequenzierung ist es möglich, den Fokus der Analyse nicht auf nur eines oder sehr wenige, sondern alle differenzialdiagnostisch bedeutsamen Gene beziehungsweise sogar das ganze Exom oder auch Genom zu legen. Die Genom- beziehungsweise Exomanalyse ist derzeit für routinediagnostische Fragestellungen aus vor allem zwei Gründen (noch) nicht geeignet: 1. Die Auswertung der generierten Daten ist extensiv und eine endgültige Beurteilung aller nachgewiesenen genetischen Varianten nicht ohne Weiteres möglich. 2. Die Abdeckung (Qualität) der Sequenz ist in vielen Bereichen des Erbguts ungenügend und erlaubt keine eindeutigen Aussagen zum Vorhandensein oder Ausschluss etwaiger Mutationen (70–72). Die Methode des «targeted next generation sequencing» beziehungsweise der Panel-Diagnostik stellt jedoch eine Möglichkeit dar, den diagnostischen Fokus ausschliesslich auf jene Gene zu legen, von welchen eine differenzialdiagnostische Bedeutung bekannt ist. Dies reduziert die Wahrscheinlichkeit des Auftretens unklarer Befunde beziehungsweise möglicher pathologischer Zufallsbefunde, die primär nicht im Zusammenhang mit der initialen Fragestellung zu stehen scheinen. Des Weiteren zeigen die Gene innerhalb eines solchen Panels eine deutlich höhere Abdeckung und erlauben gegenüber der Exom- oder Genomanalyse einen sicheren Nachweis beziehungsweise Ausschluss von Sequenzveränderungen (45). Die Aufklärungsquote einer solchen Panel-Diagnostik hängt von der Anzahl der fokussierten Gene sowie der (bekannten) Heterogenität der jeweiligen Erkrankung ab. Mittels Epilepsie-Panel wird für epileptische Enzephalopathien eine Aufklärungsrate von zirka 30 Prozent erzielt (Lemke, nicht publiziert), was mehr als dem Dreifachen der Ausbeute der Microarray-Analyse entspricht. Im Sinne einer höheren Effizienz ist daher zu diskutieren, die Panel-Diagnostik trotz derzeit höherer Kosten der Array-Diagnostik bereits vorzuschalten. Darüber hinaus zeichnet sich bereits ab, dass durch diese modernen Hochdurchsatz-Sequenziermethoden in den nächsten Jahren ein deutlicher Wissenszuwachs zu erwarten ist, der mehr Klarheit bezüglich der Ätiologie verschiedener epileptischer Enzephalopathien und vieler anderer Erkrankungen bringen wird. Korrespondenzadresse: Dr. Alexandre N. Datta Abteilung für Neuro- und Entwicklungspädiatrie Universitäts-Kinderspital Basel (UKBB) Spitalstrasse 33 4056 Basel E-Mail: Alexandre.Datta@ukbb.ch Literatur: 1. Berg AT, Berkovic SF, Brodie MJ et al.: Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010; 51: 676–685. 2. Lemke JR, Bürki SE: Genetik der infantilen epileptischen Enzephalopathien. Epileptologie 2013; 30: 5–13. 3. Ohtahara S, Ishida T, Oka E, Yamatogy Y, Inoue H: On the specific age dependent epileptic syndrome: the early-infantile epileptic encephalopathy with suppression-burst. No to Hattatsu 1976; 8: 270–279. 4. Yamatogi Y, Ohtahara S: Early-infantile epileptic encephalopathy with suppressionbursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain Dev 2002; 24: 13–23. 5. Pavone P, Spalice A, Polizzi A, Parisi P, Ruggieri M: Ohtahara syndrome with emphasis on recent genetic discovery. Brain Dev 2012; 34: 459–468. 6. Ohtahara S, Yamatogi Y: Epileptic encephalopathies in early infancy with suppressionburst. J Clin Neurophysiol 2003; 20: 398–407. 7. Kato M, Saitoh S, Kamei A et al.: A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet 2007; 81: 361–366. 8. Beal JC, Cherian K, Moshe SL: Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr Neurol 2012; 47: 317–323. 9. Uyanik G, Aigner L, Martin P et al.: ARX mutations in X-linked lissencephaly with abnormal genitalia. Neurology 2003; 61: 232–235. 10. Giordano L, Sartori S, Russo S et al.: Familial Ohtahara syndrome due to a novel ARX gene mutation. Am J Med Genet A 2010; 152A: 3133–3137. 11. Guerrini R, Moro F, Kato M et al.: Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology 2007; 69: 427–433. 12. Deprez L, Weckhuysen S, Holmgren P et al.: Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology 2010; 75: 1159–1165. 13. Weckhuysen S, Holmgren P, Hendrickx R et al.: Reduction of seizure frequency after epilepsy surgery in a patient with STXBP1 encephalopathy and clinical description of six novel mutation carriers. Epilepsia 2013; 54: e74–80. 14. Weckhuysen S, Mandelstam S, Suls A et al.: KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012; 71: 15–25. 15. Weckhuysen S, Ivanovic V, Hendrickx R et al.: Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 2013; 81: 1697–1703. 16. Aicardi J, Goutieres F: [Neonatal myoclonic encephalopathy (author's transl)]. Rev Electroencephalogr Neurophysiol Clin 1978; 8: 99–101. 17. Murakami N, Ohtsuka Y, Ohtahara S: Early infantile epileptic syndromes with suppression-bursts: early myoclonic encephalopathy vs. Ohtahara syndrome. Jpn J Psychiatry Neurol 1993; 47: 197–200. 18. Aicardi J, Ohtahara S: (2002) Severe neonatal epilepsies with suppression-burst pattern. In: Roger J, Bureau M, Dravet CH et al. (edt): Epileptic syndromes in infancy, childhood and adolescence. 3rd ed. London, John Libbey & company Ltd. 2002; 33–44. 19. Ohtahara S, Yamatogi Y: Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res 2006; 70 (Suppl 1): S58–67. 20. Backx L, Ceulemans B, Vermeesch JR, Devriendt K, Van Esch H: Early myoclonic encephalopathy caused by a disruption of the neuregulin-1 receptor ErbB4. Eur J Hum Genet 2009; 17: 378–382. 21. Coppola G, Plouin P, Chiron C, Robain O, Dulac O: Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia 1995; 36: 1017–1024. 22. Coppola G: Malignant migrating partial seizures in infancy: an epilepsy syndrome of unknown etiology. Epilepsia 2009; 50 (Suppl 5): 49–51. 23. Engel J Jr and International League Against Epilepsy: A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001; 42: 796–803. 24. Dulac O: Issues in paediatric epilepsy. Acta Neurol Scand 2005; 2 (Suppl 18): 9–11. 25. Barcia G, Fleming MR, Deligniere A et al.: De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012; 44: 1255–1259. 26. Milh M, Falace A, Villeneuve N et al.: Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy. Hum Mutat 2013; 34: 869–872. 27. Poduri A, Chopra SS, Neilan EG et al.: Homozygous PLCB1 deletion associated with malignant migrating partial seizures in infancy. Epilepsia 2012; 53: e146-150. 28. Freilich ER, Jones JM, Gaillard WD et al.: Novel SCN1A mutation in a proband with malignant migrating partial seizures of infancy. Arch Neurol 2011; 68: 665-671. 29. Coppola G, Veggiotti P, Del Giudice EM et al.: Mutational scanning of potassium, sodium and chloride ion channels in malignant migrating partial seizures in infancy. Brain Dev 2006; 28: 76-79. 26 3/14 SCHWERPUNKT 30. Panayiotopoulos C: Epileptic encephalopathies in infancy and early childhood: West syndrome. In: Panayiotopoulos C. (ed.): A Clinical Guide to Epileptic Syndromes and Their Treatment. London: Springer Verlag, 2007: 224–231. 31. Nabbout R, Dulac O: Epileptic encephalopathies: a brief overview. J Clin Neurophysiol 2003; 20: 393–397. 32. Paciorkowski AR, Thio LL, Dobyns WB: Genetic and biologic classification of infantile spasms. Pediatr Neurol 2011; 45: 355–367. 33. Kurian MA, Meyer E, Vassallo G et al.: Phospholipase C beta 1 deficiency is associated with early-onset epileptic encephalopathy. Brain 2010; 133: 2964–2970. 34. Edvardson S, Baumann AM, Muhlenhoff M et al.: West syndrome caused by ST3GalIII deficiency. Epilepsia 2013; 54: e24–27. 35. Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P et al.: De novo mutations in epileptic encephalopathies. Nature 2013; 501: 217–221. 36. Lemke JR, Hendrickx R, Geider K et al.: GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol 2014; 75: 147–154. 37. Dravet C: Les épilepsies graves de l'enfant. Vie Med 1978; 8: 543–548. 38. Scheffer IE: Diagnosis and long-term course of Dravet syndrome. Eur J Paediatr Neurol 2012; 16 (Suppl 1), S5–8. 39. Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM: Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 2012; 135: 2329–2336. 40. Claes L, Del-Favero J, Ceulemans B et al.: De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001; 68: 1327–1332. 41. Depienne C, Bouteiller D, Keren B et al.: Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 2009; 5: e1000381. 42. Suls A, Jaehn JA, Kecskes A et al.: De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 2013, 93: 967–975. 43. Carvill GL, Weckhuysen S, McMahon JM et al.: GABRA1 and STXBP1: Novel genetic causes of Dravet syndrome. Neurology 2014; 82(14): 1245–1253. 44. Singh NA, Pappas C, Dahle EJ et al.: A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet 2009; 5: e1000649. 45. Lemke JR, Riesch E, Scheurenbrand T et al.: Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012; 53: 1387–1398. 46. Lennox WG, Davis JP: Clinical correlates of the fast and the slow spike-wave electroencephalogram. Pediatrics 1950; 5: 626–644. 47. Gastaut H: Certain Basic Concepts Concerning the Treatment of the Epilepsies. Br J Clin Pract 1964; 18: 463–468. 48. Eriksson KJ, Koivikko MJ: Prevalence, classification, and severity of epilepsy and epileptic syndromes in children. Epilepsia 1997; 38: 1275–1282. 49. Berg AT, Shinnar S, Levy SR, Testa FM: Newly diagnosed epilepsy in children: presentation at diagnosis. Epilepsia 1999; 40: 445–452. 50. Baumanoir A, Blume W: The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet C et al.: Epileptic syndromes in Infancy, Childhood, and Adolescence. Libbey Eurotext 2012; 125. 51. Kaminska A, Oguni H: Lennox-Gastaut syndrome and epilepsy with myoclonic-astatic seizures. In: Dulac O, Lassonde M, Sarnat HB: Handbook of Clinical Neurology 2013; 111: 641–652. 52. Schmitt B, Wohlrab G: EEG in der Neuropädiatrie. In: Zschocke S, Hansen HC: Klinische Elektroencephalographie 2012; 3: 523–586. 53. Lund C, Brodtkorb E, Oye AM, Rosby O, Selmer KK: CHD2 mutations in LennoxGastaut syndrome. Epilepsy Behav 2014; 33C: 18–21. 54. Kruse R: [The myoclonic astatic petit mal. Clinical course of small epileptic seizures in childhood. With an introduction by Prof. Dr. Dietrich Janz]. Monogr Gesamtgeb Neurol Psychiatr 1968; 124: 1–126. 55. Doose H, Gerken H, Leonhardt R, Volzke E, Volz C: Centrencephalic myoclonic-astatic petit mal. Clinical and genetic investigation. Neuropädiatrie 1970; 2: 59–78. 56. Oguni H, Hayashi K, Imai K, Funatsuka M, Sakauchi M, Shirakawa S, Osawa M: Idiopathic myoclonic-astatic epilepsy of early childhood--nosology based on electrophysiologic and long-term follow-up study of patients. Adv Neurol 2005; 95: 157–174. 57. Patry G, Lyagoubi S, Tassinari CA: Subclinical «electrical status epilepticus» induced by sleep in children. A clinical and electroencephalographic study of six cases. Arch Neurol 1971; 24: 242–252. 58. Engel J Jr, Berg A, Andermann F et al.: Are epilepsy classifications based on epileptic syndromes and seizure types outdated? Epileptic Disord 2006; 8: 159–160. 59. Datta AN, Oser N, Ramelli GP, Gobbin NZ, Lantz G, Penner IK, Weber P: BECTS evolving to Landau-Kleffner Syndrome and back by subsequent recovery: a longitudinal language reorganization case study using fMRI, source EEG, and neuropsychological testing. Epilepsy Behav 2013; 27: 107–114. 60. Scheltens-de Boer M: Guidelines for EEG in encephalopathy related to ESES/CSWS in children. Epilepsia 2009; 50 (Suppl 7): 13-17. 61. Tassinari CA, Rubboli G, Volpi L et al.: Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin Neurophysiol 2000; 111 (Suppl 2): S94-S102. 62. Heijbel J, Blom S, Rasmuson M: Benign epilepsy of childhood with centrotemporal EEG foci: a genetic study. Epilepsia 1975; 16: 285–293. 63. Degen R, Degen HE: Contribution to the genetics of rolandic epilepsy: waking and sleep EEGs in siblings. Epilepsy Res 1992; Suppl 6: 49–52. 64. Lemke JR, Lal D, Reinthaler EM et al.: Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 2013; 45: 1067–1072. 65. Lesca G, Rudolf G, Bruneau N et al.: GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 2013; 45: 1061–1066. 66. Carvill GL, Regan BM, Yendle SC et al.: GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet 2013; 45: 1073–1076. 67. Rauch A, Wieczorek D, Graf E et al.: Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012; 380: 1674–1682. 68. Deprez L, Jansen A, De Jonghe P: Genetics of epilepsy syndromes starting in the first year of life. Neurology 2009; 72: 273–281. 69. Mefford HC, Yendle SC, Hsu C et al.: Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol 2011; 70: 974–985. 70. Ng SB, Turner EH, Robertson PD et al.: Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009; 461: 272–276. 71. 1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A et al.: A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073. 72. Scheffer IE: Genetic testing in epilepsy: what should you be doing? Epilepsy Curr 2011; 11: 107–111. 3/14 27