



Transkript
Im Fokus: Leukämien bei Erwachsenen und Kindern
Akute Leukämien im Kindesalter
Inzidenz, Ätiologie, Diagnostik, Therapiemodalitäten, Prognose
Die heutige Behandlung der akuten Leukämien erfolgt risikoadaptiert, basierend auf verschiedenen biologischen Parametern. Leukämiesubtyp und zytogenetische Veränderungen bei Diagnose sowie die Dynamik des Leukämierückganges nach Therapieinduktion spielen für die Therapieintensität eine dominante Rolle. Damit konnten die Heilungsraten sowohl bei ALL wie bei AML in den letzten Jahren weiter verbessert werden.
FELIX K. NIGGLI, NICOLE BODMER
Felix K. Niggli
Nicole Bodmer
In industrialisierten Ländern ist die Leukämie mit zirka 35% aller Neoplasien die häufigste Krebserkrankung im Kindesalter. In der Schweiz werden jährlich 55 bis 65 Neuerkrankungen diagnostiziert. Die aktuelle Inzidenz bei Kindern unter 15 Jahren liegt bei etwa 4,9 Fälle/100 000 Einwohner. Die zwei wichtigsten Formen der Leukämie sind ▲ die akute lymphatische Leukämie (ALL) (ca. 82%)
sowie ▲ die akute myeloische Leukämie (AML) (ca. 15%). Selten treten myelodysplastische Syndrome sowie chronische myeloische Leukämien (CML) bei Kindern auf.
Akute lymphatische Leukämie (ALL)
Die ALL entwickelt sich aus einer unreifen Zelle der B- oder der T-Zellreihe des lymphatischen Systems. Am häufigsten kommt es zur B-Vorläufer-ALL. Eine Sonderform ist die reife B-Zell-Leukämie, die auf einer malignen Transformation der reifen B-Zelle beruht und als leukämische Manifestation eines BurkittLymphoms verstanden wird.
Ätiologie Die Ursache der Leukämie ist nach wie vor unklar. Bekannte Faktoren sind ionisierende Strahlung sowie kongenitale Syndrome. Damit werden aber weniger als 10% aller Erkrankungen erklärt. Man schätzt, dass zirka 1% der Kinder, welche mit Down-Syndrom ge-
ABSTRACT
Leukaemia in childhood
Acute leukaemia is the most common cancer in children. Continuous progress in treatment and a better understanding of the biology of this disease has pushed the cure rate in acute lymphoblastic leukaemia to more than 85% and in acute myeloid leukaemia up to 70%. Today, molecular techniques contribute significantly to the initial risk stratification and response assessment and are important for therapy adjustment. Current efforts aim to develop more effective strategies for prognostic unfavourable high risk leukaemia and to reduce toxicities of therapy in favourable subgroups.
Keywords: leukaemia, treatment, chemotherapy, bone marrow.
SZO 2013; 2: 12–16.
boren werden, innerhalb der ersten 5 Jahre eine Leukämie entwickelt (Risiko etwa 20-fach vergrössert), wobei sowohl eine ALL wie auch eine AML auftreten kann. Noch deutlich häufiger (bei 3–10%) tritt bei diesen Kindern eine transiente Myeloproliferation im Neugeborenenalter auf, welche gelegentlich später in eine Leukämie übergeht. Weitere seltenere angeborene Veränderungen mit erhöhtem Leukämierisiko sind Ataxia teleangiectatica, Fanconi-Syndrom und andere Syndrome, welche mit Immundefizienz oder erhöhter Chromosomenbrüchigkeit einhergehen. Die Tatsachen, dass ALL gehäuft zwischen dem 2. und 5. Lebensjahr auftritt, eine Häufung der Erkrankung in industrialisierten Ländern zu erkennen ist, sowie die Beobachtung, dass es in der Vergangenheit immer wieder zu Clusterbildung speziell in Regionen neuer Agglomerationen gekommen ist, führte zu zwei möglichen infektassoziierten Hypothesen der Leukämieentstehung: ▲ Die Kinlen-Hypothese («Bevölkerungsdurchmi-
schungshypothese») geht davon aus, dass Leukämiecluster das Resultat einer Exposition empfindlicher, nicht immuner Individuen gegenüber häufigen, kaum pathogenen Infektionserregern sein könnte, die sich im Rahmen von Bevölkerungsdurchmischung verbreiten (1). ▲ Die Greaves-Hypothese («verzögerter Infektionskontakt») der Leukämieentstehung basiert auf einer verspäteten Immunantwort gegenüber typischen Infektionserregern, welche zur Ätiologie der B-Vorläufer-ALL beitragen könnte (2). Dabei wird vermutet, dass empfindliche Individuen, welche bereits mit einem präleukämischen Klon geboren wurden und welche im frühen Lebensalter nur geringfügig mit Infekterregern in Kontakt kamen, schliesslich verspätet (bei entsprechender Konfrontation mit Infektionserregern) zu abnormen Immunreaktionen mit pathologischer Lymphoproliferation befähigt sind. Chromosomenveränderungen, wie Mutationen und Deletionen, welche sich schliesslich oft in Transloka-
12 SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 2/2013
Im Fokus: Leukämien bei Erwachsenen und Kindern
tionen oder Hyperdiploidien äussern, sind oft frühe initiierende Ereignisse einer Leukämieentwicklung, welche sich meist bereits pränatal entwickeln (3). Solche Aberrationen konnten mehrfach retrospektiv in archivierten, bei der Geburt entnommenen Blutproben von Kindern mit Leukämien nachgewiesen werden. Da die Inzidenz einer manifesten Leukämie aber deutlich geringer ist als der Nachweis solcher genetischer Veränderungen bei gesunden Neugeborenen, ist es wahrscheinlich, dass weitere Ereignisse zur Leukämieentstehung beitragen. Ein solches Modell für eine Leukämieentstehung ist in Abbildung 1 dargestellt. Sowohl Art und Weise der Mutation wie auch der Zeitpunkt ihres Auftretens sind vermutlich entscheidend für die daraus entstehende klonale Proliferation.
Klinik Bei der ALL stehen Symptome, welche durch die Unterdrückung der normalen Blutbildung verursacht sind, im Vordergrund. Durch Infiltrationen kommt es gehäuft zu diffusen Knochenschmerzen und wechselnden Arthropathien, welche sich bei Kleinkindern gelegentlich als Bewegungsunlust bis zur Gehverweigerung äussern. Im Weiteren kann es zu Lymphknotenschwellungen sowie Organomegalien kommen.
Diagnostik Im Blut findet man primär am häufigsten eine Thrombozytopenie und eine Anämie. Die Leukozytenzahl kann normal erniedrigt oder erhöht sein. Diagnostisch konklusiv ist üblicherweise die Morphologie aus einer Knochenmarkpunktion. Daraus wird auch der Immunphänotyp mittels Durchflusszytometrie (FACS) bestimmt sowie eine Chromosomenanalyse durchgeführt. Die Immunphänotypisierung ermöglicht die Bestimmung des Entwicklungsstadiums des entsprechenden Zellklons. Der häufigste Leukämiesubtyp bei Kindern, die sogenannte «common ALL», ist durch Expression der B-Zell-Marker CD10 und CD 19 charakterisiert. Eine Expression myeloischer Antigene – welche meist jedoch prognostisch nicht bedeutungsvoll ist – kann fast in der Hälfte der ALLFälle nachgewiesen werden. Einen wichtigen Stellenwert hat heutzutage die zytogenetische und molekulargenetische Diagnostik. Dabei gilt es, die wichtigsten Subgruppen zu erkennen, da sie therapeutische Implikationen haben. Einerseits werden numerische Veränderungen wie Hyperdiploidie, Hypodiploidie sowie strukturelle Veränderungen wie Translokationen, zum Beispiel t(12; 21) (Fusion der Gene ETV6/RUNX1) oder t(9;22) (Fusion von BCR/ABL1), MLL-Rearrangements (MLL 11q23) und andere Veränderungen gesucht. Klassisch erfolgt der Nachweis dieser Veränderungen mittels konventioneller Zytogenetik (G-Banding) und/oder fluoreszierender In-situ-Hybridisierung (FISH) in den Leukämiezel-
Abbildung 1: Zweistufenmodell der Leukämieentstehung. Eine erste Mutation in einer hämatopoetischen Stammzelle induziert einen präleukämischen Klon. Dieser bleibt meist vorerst stumm. Ein weiteres postnatales Ereignis, zum Beispiel eine Infektion, könnte in einem präleukämischen Klon eine Leukämie auslösen.
len. Die häufigsten chromosomalen Veränderungen bei ALL sind in Tabelle 1 wiedergegeben. Im Rahmen der Verlaufsdiagnostik von Leukämien hat sich in den vergangenen Jahren die Messung der minimalen Resterkrankung (minimal residual disease, MRD) aus dem Knochenmark etabliert. Dabei werden heute im Wesentlichen zwei Methoden angewendet, die sich im klinischen Alltag ergänzen. Die empfindlichste Methode ist das Monitoring von Immunglobulin- und T-Zell-Rezeptor-Rearrangements. Dabei werden initial leukämiespezifische klonale Rearrangements gesucht, die mittels quantitativer PCR zu bestimmten Therapiezeitpunkten verfolgt werden. Die damit erreichte Detektionsgrenze liegt bei zirka 1 Leukämiezelle unter 100 000 normalen Knochenmarkzellen. Eine um etwa eine Logstufe weniger sensitive Technik der MRD-Messung beruht auf dem Monitoring des leukämieassoziierten Immunphänotyps mittels Durchflusszytometrie. Dabei kann eine Sensitivität von 0,001% erreicht werden (4).
Therapie und Prognose Die kontinuierliche Anpassung der Behandlung der ALL während der letzten 40 Jahre hat zu einem dramatischen Anstieg der Überlebenswahrscheinlichkeit geführt. Die ALL-BFM-Studiengruppe hat seit 1976 in zahlreichen gross angelegten, randomisierten Therapiestudien wesentlich zu dieser Therapieverbesserung beigetragen. Durch eine Induktionstherapie sollen eine Remission und eine Wiederherstellung einer normalen Hämatopoese innert 4 bis 6 Wochen erreicht werden. Dies ist mit den heutigen Therapiemodalitäten bei zirka 98% aller Patienten mit den drei Medikamenten Glukokortikoide, Vincristin und Asparaginase möglich, wobei je nach Studiengruppe noch ein Anthrazyklin hinzugefügt wird (5, 6). In ALLBFM-Protokollen wird die Remissionsinduktion von einer 7-tägigen Monotherapie mit Prednison sowie einer intrathekalen Verabreichung von Methotrexat eingeleitet. Damit können mehrheitlich Komplikationen durch Zellzerfall vermieden werden.
SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 2/2013
13
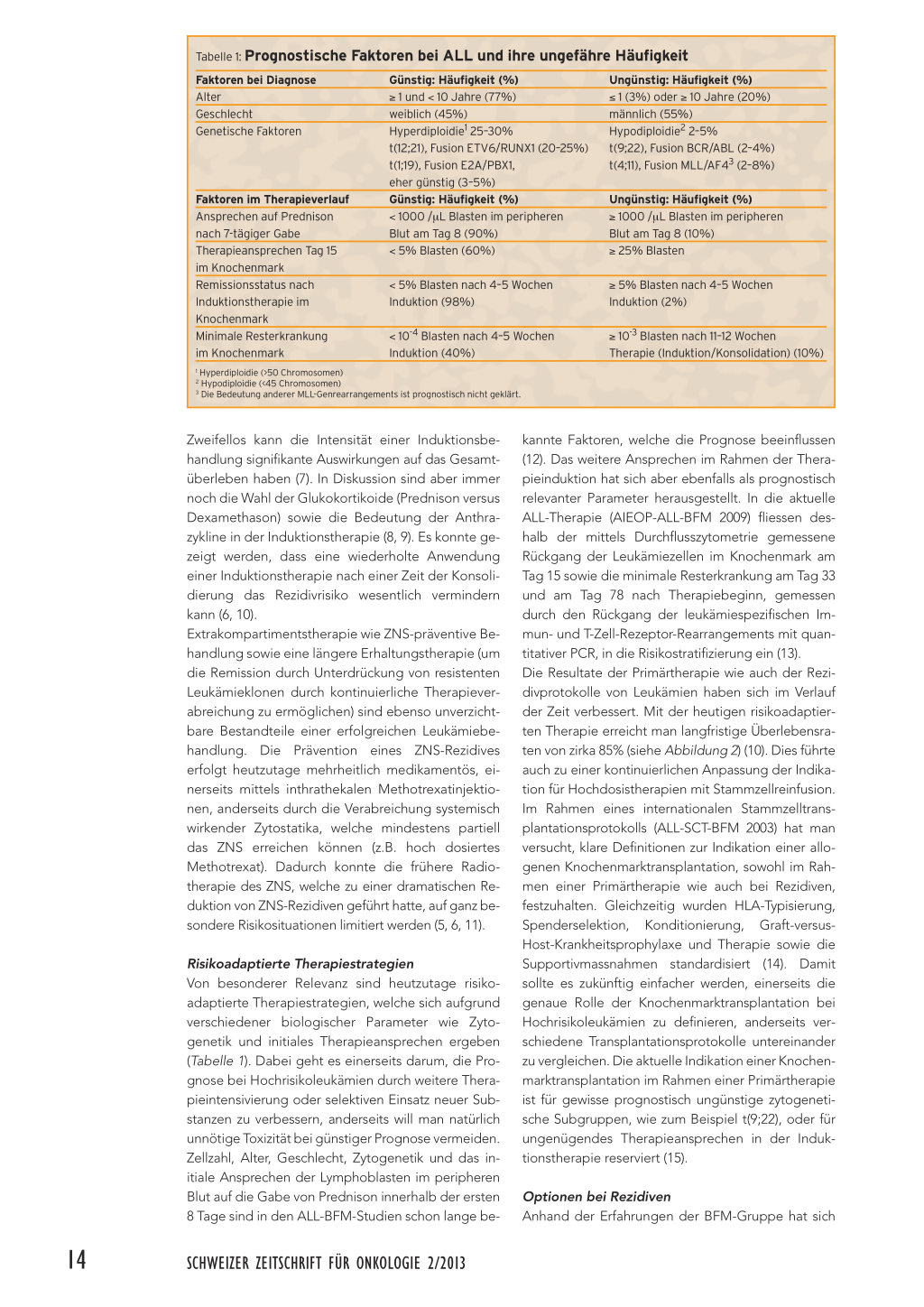
Tabelle 1: Prognostische Faktoren bei ALL und ihre ungefähre Häufigkeit
Faktoren bei Diagnose Alter Geschlecht Genetische Faktoren
Faktoren im Therapieverlauf Ansprechen auf Prednison nach 7-tägiger Gabe Therapieansprechen Tag 15 im Knochenmark Remissionsstatus nach Induktionstherapie im Knochenmark Minimale Resterkrankung im Knochenmark
Günstig: Häufigkeit (%) ≥ 1 und < 10 Jahre (77%) weiblich (45%) Hyperdiploidie1 25–30% t(12;21), Fusion ETV6/RUNX1 (20–25%) t(1;19), Fusion E2A/PBX1, eher günstig (3–5%) Günstig: Häufigkeit (%) < 1000 /μL Blasten im peripheren Blut am Tag 8 (90%) < 5% Blasten (60%) < 5% Blasten nach 4–5 Wochen Induktion (98%) < 10-4 Blasten nach 4–5 Wochen Induktion (40%) 1 Hyperdiploidie (>50 Chromosomen) 2 Hypodiploidie (<45 Chromosomen) 3 Die Bedeutung anderer MLL-Genrearrangements ist prognostisch nicht geklärt. Ungünstig: Häufigkeit (%) ≤ 1 (3%) oder ≥ 10 Jahre (20%) männlich (55%) Hypodiploidie2 2–5% t(9;22), Fusion BCR/ABL (2–4%) t(4;11), Fusion MLL/AF43 (2–8%) Ungünstig: Häufigkeit (%) ≥ 1000 /μL Blasten im peripheren Blut am Tag 8 (10%) ≥ 25% Blasten ≥ 5% Blasten nach 4–5 Wochen Induktion (2%) ≥ 10-3 Blasten nach 11–12 Wochen Therapie (Induktion/Konsolidation) (10%) Zweifellos kann die Intensität einer Induktionsbehandlung signifikante Auswirkungen auf das Gesamtüberleben haben (7). In Diskussion sind aber immer noch die Wahl der Glukokortikoide (Prednison versus Dexamethason) sowie die Bedeutung der Anthrazykline in der Induktionstherapie (8, 9). Es konnte gezeigt werden, dass eine wiederholte Anwendung einer Induktionstherapie nach einer Zeit der Konsolidierung das Rezidivrisiko wesentlich vermindern kann (6, 10). Extrakompartimentstherapie wie ZNS-präventive Behandlung sowie eine längere Erhaltungstherapie (um die Remission durch Unterdrückung von resistenten Leukämieklonen durch kontinuierliche Therapieverabreichung zu ermöglichen) sind ebenso unverzichtbare Bestandteile einer erfolgreichen Leukämiebehandlung. Die Prävention eines ZNS-Rezidives erfolgt heutzutage mehrheitlich medikamentös, einerseits mittels inthrathekalen Methotrexatinjektionen, anderseits durch die Verabreichung systemisch wirkender Zytostatika, welche mindestens partiell das ZNS erreichen können (z.B. hoch dosiertes Methotrexat). Dadurch konnte die frühere Radiotherapie des ZNS, welche zu einer dramatischen Reduktion von ZNS-Rezidiven geführt hatte, auf ganz besondere Risikosituationen limitiert werden (5, 6, 11). Risikoadaptierte Therapiestrategien Von besonderer Relevanz sind heutzutage risikoadaptierte Therapiestrategien, welche sich aufgrund verschiedener biologischer Parameter wie Zytogenetik und initiales Therapieansprechen ergeben (Tabelle 1). Dabei geht es einerseits darum, die Prognose bei Hochrisikoleukämien durch weitere Therapieintensivierung oder selektiven Einsatz neuer Substanzen zu verbessern, anderseits will man natürlich unnötige Toxizität bei günstiger Prognose vermeiden. Zellzahl, Alter, Geschlecht, Zytogenetik und das initiale Ansprechen der Lymphoblasten im peripheren Blut auf die Gabe von Prednison innerhalb der ersten 8 Tage sind in den ALL-BFM-Studien schon lange be- kannte Faktoren, welche die Prognose beeinflussen (12). Das weitere Ansprechen im Rahmen der Therapieinduktion hat sich aber ebenfalls als prognostisch relevanter Parameter herausgestellt. In die aktuelle ALL-Therapie (AIEOP-ALL-BFM 2009) fliessen deshalb der mittels Durchflusszytometrie gemessene Rückgang der Leukämiezellen im Knochenmark am Tag 15 sowie die minimale Resterkrankung am Tag 33 und am Tag 78 nach Therapiebeginn, gemessen durch den Rückgang der leukämiespezifischen Immun- und T-Zell-Rezeptor-Rearrangements mit quantitativer PCR, in die Risikostratifizierung ein (13). Die Resultate der Primärtherapie wie auch der Rezidivprotokolle von Leukämien haben sich im Verlauf der Zeit verbessert. Mit der heutigen risikoadaptierten Therapie erreicht man langfristige Überlebensraten von zirka 85% (siehe Abbildung 2) (10). Dies führte auch zu einer kontinuierlichen Anpassung der Indikation für Hochdosistherapien mit Stammzellreinfusion. Im Rahmen eines internationalen Stammzelltransplantationsprotokolls (ALL-SCT-BFM 2003) hat man versucht, klare Definitionen zur Indikation einer allogenen Knochenmarktransplantation, sowohl im Rahmen einer Primärtherapie wie auch bei Rezidiven, festzuhalten. Gleichzeitig wurden HLA-Typisierung, Spenderselektion, Konditionierung, Graft-versusHost-Krankheitsprophylaxe und Therapie sowie die Supportivmassnahmen standardisiert (14). Damit sollte es zukünftig einfacher werden, einerseits die genaue Rolle der Knochenmarktransplantation bei Hochrisikoleukämien zu definieren, anderseits verschiedene Transplantationsprotokolle untereinander zu vergleichen. Die aktuelle Indikation einer Knochenmarktransplantation im Rahmen einer Primärtherapie ist für gewisse prognostisch ungünstige zytogenetische Subgruppen, wie zum Beispiel t(9;22), oder für ungenügendes Therapieansprechen in der Induktionstherapie reserviert (15). Optionen bei Rezidiven Anhand der Erfahrungen der BFM-Gruppe hat sich 14 SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 2/2013 Im Fokus: Leukämien bei Erwachsenen und Kindern gezeigt, dass der Behandlungserfolg bei Rezidiven vor allem vom Zeitpunkt des Auftretens eines Rezidivs, vom Befallsmuster der Leukämie sowie vom Leukämiesubtyp abhängt (16). Aber auch hier wurde gezeigt, dass das Therapieansprechen nach erneuter Therapieinduktion und somit die Dynamik des Rückgangs der minimalen Resterkrankung von besonderer prognostischer Bedeutung sind und entsprechend weitere Therapieelemente (wie der Einsatz einer Knochenmarktransplantation) danach ausgerichtet werden können (17). Es besteht berechtigte Hoffnung, dass zukünftig eine zielgerichtete Therapie mit Antikörpern oder spezifischeren Kinaseinhibitoren bei verschiedenen Subtypen von ALL möglich sein wird. Als Beispiel sei der CD19-Antikörper (Blinatumomab) genannt, der in Phase-I- und -II-Studien bei rezidivierter ALL erfreuliche Remissionsraten induzieren kann. Weitere mögliche Therapieansätze zielgerichteter Therapien beinhalten Interaktionen mit Signalwegen innerhalb der Leukämiezellen. Dazu gehört zum Beispiel die Unterdrückung der CRLF2Genexpression oder einer abnormen JAK-Gen-Aktivierung. Akute myeloische Leukämie (AML) Bei der AML ist eine myeloische Stamm- oder Vorläuferzelle betroffen, die für die Bildung von Granulozyten, Monozyten, Thrombozyten oder Erythrozyten verantwortlich ist. Bei der AML ist im Gegensatz zur ALL die immunologische Subtypisierung von geringerer Bedeutung. Zudem sind bei der AML neben dem morphologischen Subtyp genetische Faktoren sowie das Ansprechen auf die Therapie und das Monitoring der Resterkrankung wichtige Parameter in der prognostischen Bewertung (18). Translokationen wie t(8;21) (vor allem beim AML-Subtyp FAB M1/M2 vorkommend), t(15;17) (typisch für AML FAB M3) sowie Inversion inv (16) (typisch bei AML FAB M4 Eo) sind günstige Faktoren, während gewisse Subtypen mit MLL-Rearrangement (11q23) oder FLT3-Gen-Mutation prognostisch ungünstig sind (Tabelle 2) (19). Abbildung 2: Gesamtüberleben nach 10 Jahren in den verschiedenen ALLBFM Studien (10). Therapiestrategien Für die ALL scheint es keine Einheitstherapie für alle Subgruppen zu geben. Während bei der akuten Promyelozytenleukämie (AML, FAB M3) die Einführung von Retinolsäure die Komplikationsrate deutlich verringert hat und die Heilungsrate verbesserte, sind andere Subtypen ohne Knochenmarktransplantation kaum zu beherrschen. Eine Hyperleukozytose bei AML sowie der Subtyp AML, FAB M3 sind als Notfall zu betrachten und benötigen eine rasche Intervention, da das Blutungsrisiko in solchen Situation deutlich erhöht ist. Die entscheidenden Medikamente für die Remissionsinduktion bei der AML sind Cytarabin und Anthrazykline (20, 21). Oft wird auch Etoposid dazugegeben. Der zusätzliche Nutzen einer Postinduktionschemotherapie, wiederum mit hoch dosiertem Cytarabin, wurde mehrfach gezeigt. In den letzten Jahren wurde bei Hochrisikoleukämien zusätzlich 2-CDA (2-Chloro-2-Deoxyadenosin) erfolgreich eingesetzt, vor allem in Kombination mit Cytarabin. Diese Kombination führt zu einer maximalen Inhibition der DNA-Synthese. Die Erhaltungstherapie bei AML ist immer noch umstritten (22). Das Vorgehen der ZNS-gerichteten Therapie ist ebenfalls uneinheitlich. Sie reicht von intrathekaler Verabreichung einer Monotherapie mit Cytarabin oder Methotrexat bis hin zu Trippeltherapie mit Cytarabin, Methotrexat und Hydrokortison, mit oder ohne zusätzliche Radiotherapie. Tabelle 2: Biologische Faktoren bei AML und ihre prognostische Bedeutung (19) Genetische/biologische Faktoren t(8;21) inv (16) t(15;17) t(1;11) t(9;11) t(10;11) t(4;11) t(6;11) del 7q FLT3-Mutationen c-KIT Sekundäre AML Häufigkeit (%) 12–14% 8% 6–10% 3% 10% Bedeutung günstig günstig günstig günstig intermediär bis günstig ungünstig ungünstig ungünstig ungünstig ungünstig unklar ungünstig SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 2/2013 15 Im Fokus: Leukämien bei Erwachsenen und Kindern Generell wurde aber die Radiotherapie in den letzten Jahren auch bei der AML deutlich reduziert oder sogar ganz weggelassen. Die Heilungsrate der AML hängt stark von den genannten Parametern ab, erreicht aber heute bis zu 70%. Die allogene Stammzelltransplantation wird in gewissen Studiengruppen bei Vorhandensein eines kompatiblen Geschwisterspenders immer noch grosszügig empfohlen. Andere wiederum bevorzugen eine risikoadaptierte Indikation für Stammzelltransplantation. Generell ist aber eine vermehrte Zurückhaltung in der Empfehlung einer Stammzelltransplantation zu erkennen. Sicherlich ist eine solche bei unvollständiger Remission oder bei Rezidiven vorzusehen. Hoffnung wird auf den gezielten Einsatz von Kinaseinhibitoren bei nachgewiesenen Mutationen, wie FLT3-Mutationen oder KIT-Mutationen, gesetzt. Spätfolgen Mit zunehmender Überlebensrate nach Leukämien im Kindesalter wurde natürlich auch die Aufmerk- samkeit auf Spätfolgen erhöht. Zum Beispiel muss die Kardiotoxizität bei myeloischer Leukämiebe- handlung langfristig beachtet werden. Osteonekro- sen und andere vaskuläre Ereignisse, welche gehäuft bei ALL anzutreffen sind, können ebenfalls Langzeit- morbidität hervorrufen. Nach einer Knochenmark- transplantation sind sicher die Spätfolgen häufiger und relevanter. Das Risiko von Sekundärmalignomen ist wiederum abhängig von der Art der Behandlung und sicherlich von gewisser Relevanz bei Anwendung einer Radiotherapie. ▲ Prof Dr. med. Felix Niggli (Korrespondenzadresse) E-Mail: felix.niggli@kispi.uzh.ch Dr. med. Nicole Bodmer E-Mail: nicole.bodmer@kispi.uzh.ch Abteilung Onkologie Universitäts-Kinderspital Zürich Steinwiesstrasse 75 8032 Zürich Quellen: 1. Kinlen L.: Infections and immune factors in cancer: the role of epidemiology. Oncogene 2004; 23: 60–75. 2. Greaves M.: Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer 2006; 6(3): 193–203. 3. Greaves MF, Wiemels J.: Origins of chromosome translocations in childhood leukaemia. Nature Reviews Cancer 2003; 3: 639–649. 4. Campano D.: Minimal residual disease monitoring in childhood acute lymphoblastic leukemia. Curr Opin Hematol 2012; 19: 313–18. 5. Pui CH, Evans WE.: Treatment of childhood acute lymphoblastic leukemia. N Engl J Med 2006; 354: 166–78. 6. Möricke A, Reiter A, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 2008; 111(9): 4477–89. 7. Schrappe M, Reiter A, et al.: Long-term results of four consecutive trials in childhood ALL performed by the ALL-BFM study group from 1981 to 1995. Berlin-Frankfurt-Münster. Leukemia 2000; 14(12): 2205–22. 8. Bostrom BC, Sensel MR, et al.: Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with Merkpunkte ▲ Einen wichtigen Stellenwert in der Leukämiebe- handlung hat die zytogenetische und molekulargene- tische Diagnostik für die Bestimmung des Subtyps und des Therapiemonitorings. ▲ Die Leukämie im Kindesalter wird risikoadaptiert behandelt und ist in der Mehrzahl der Fälle heilbar. ▲ Die Bestimmung der minimalen Resterkrankung nach Therapieinduktion bei akuter lymphatischer Leukämie ist ein wichtiger prognostischer Faktor. ▲ Aktuelle Entwicklungen zielen auf eine effektivere Behandlung bisher resistenter Leukämiesubtypen so- wie auf eine Verringerung der Therapietoxizität. standard-risk acute lymphoblastic leukemia: a report from the Children’s Cancer Group. Blood 2003; 101(10): 3809–17. 9. Mitchell CD, Richards SM, et al.: Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 2005; 129(6): 734–45. 10. Möricke A, Zimmermann M, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 2010; 24(2): 265–84. 11. Kamps WA, Bökkerink JPet al.: BFM-oriented treatment for children with acute lymphoblastic leukemia without cranial irradiation and treatment reduction for standard risk patients: results of DCLSG protocol ALL8 (1991–1996). Leukemia 2002; 16(6): 1099–111. 12. Schrappe M, Reiter A, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALLBFM Study Group. Blood 2000; 95(11): 3310–22. 13. International Collaborative Treatment Protocol For Children And Adolescents With Acute Lymphoblastic Leukemia. http: //clinicaltrials.gov/show/NCT01117441. 14. Schrauder A, von Stackelberg A, et al.: ALL-BFM Study Group; EBMT PD WP; I-BFM Study Group. Allogeneic hematopoietic SCT in children with ALL: current concepts of ongoing prospective SCT trials. Bone Marrow Transplant 2008; 41 Suppl 2: S71–4. 15. Balduzzi A, Valsecchi MG, et al.: Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 2005; 366: 635–42. 16. Tallen G, Ratei R, et al.: Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol 2010; 28: 2339–47. 17. Eckert C, von Stackelberg A, et al.: Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia – Long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 2012; http://dx.doi.org/10.1016/j.ejca. 2012.11.010. 18. Abrahamsson J, Forestier E, et al.: Response-guided induction therapy in pediatric acute myeloid leukemia with excellent remission rate. J Clin Oncol 2011; 29(3): 310–15. 19. Creutzig U, van den Heuvel-Eibrink MM, et al.: AML Committee of the International BFM Study Group. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 2012; 120(16): 3187–205. 20. Creutzig U, Zimmermann M, et al.: Treatment strategies and longterm results in paediatric patients treated in four consecutive AML-BFM trials. Leukemia 2005; 19(12): 2030–42. 21. Creutzig U, Zimmermann M, et al.: Less toxicity by optimizing chemotherapy, but not by addition of granulocyte colony-stimulating factor in children and adolescents with acute myeloid leukemia: results of AMLBFM 98. J Clin Oncol 2006; 24(27): 4499–506. 22. Perel Y, Auvrignon A, et al.: Group LAME of the French Society of Pediatric Hematology and Immunology. Impact of addition of maintenance therapy to intensive induction and consolidation chemotherapy for childhood acute myeloblastic leukemia: results of a prospective randomized trial, LAME 89/91. J Clin Oncol 2002; 20(12): 2774–82. 16 SCHWEIZER ZEITSCHRIFT FÜR ONKOLOGIE 2/2013