



Transkript
Im Fokus: Tumoren im Kindesalter
Knochen- und Weichteilsarkome im Jugendalter
Klinik, Diagnostik,Therapien und Prognosen
Sarkome sind nach Leukämien, Hirntumoren und Lymphomen die vierthäufigste Tumorgruppe bei Kindern. Knochen- und Weichteilsarkome stellen eine diagnostische und therapeutische Herausforderung dar und erfordern ein interdisziplinäres Team für das optimale Management. Erscheinungsformen, diagnostische Interventionen, Therapiemöglichkeiten und Prognosen werden im Folgenden dargestellt.
NICOLE BODMER, FELIX K. NIGGLI
Felix K. Niggli
14
Bösartige Knochentumore stehen in der zweiten Lebensdekade, also bei Kindern und Jugendlichen zwischen 10 und 20 Jahren, an 2. bis 3. Stelle aller bösartigen Erkrankungen. Osteosarkome (OS) stellen mit 60% die häufigsten malignen Tumore des Skelettsystems dar, gefolgt von Ewing-Sarkomen, die für weitere 25% verantwortlich sind. Die jährliche Inzidenz für maligne Knochentumoren liegt zwischen 0,5 und 2/100 000.
Osteosarkom
80% der OS treten typischerweise an den Metaphysen der langen Röhrenknochen auf, wobei am häufigsten der distale Femur betroffen ist, gefolgt von der proximalen Tibia und dem proximalen Humerus. Viel seltener ist ein Befall des Achsenskeletts. Es besteht eine deutliche Knabenwendigkeit mit einem Geschlechterverhältnis von 3:2. Mehr als 80% der Patienten haben bei Diagnose eine lokalisierte Erkrankung, die übrigen Patienten sind am häufigsten von Lungenmetastasen betroffen; Knochenmetastasen sind sehr viel seltener. Die Ätiologie ist unbekannt, neben genetischer Prädisposition können exogene Faktoren wie ionisierende Strahlen und alkylierende Substanzen eine Rolle spielen. Bei etwa 3% der betroffenen Patienten findet sich eine Veränderung des Tumorsuppressorgens p53 (1, 2).
Klinische Präsentation Häufig kommt es erst zur verzögerten Diagnosestellung eines OS, weil die Symptome lange unspezifisch sein können. Nicht selten erstreckt sich die Anamnesedauer daher über Monate. Am meisten werden Schmerz, Schwellung und Überwärmung beobachtet (2). Bei etwa 5 bis 10% der Patienten sind pathologi-
sche Frakturen das erste Symptom (3). Allgemeinsymptome sind generell selten, und die Labordiagnostik ist in der Regel normal bis auf Erhöhungen der Laktatdehydrogenase (LDH) oder der alkalischen Phosphatase (4).
Bildgebende Untersuchungen Der erste Schritt der diagnostischen Abklärungen besteht in einem konventionellen Röntgenbild, das neben destruktiven Knochenläsionen häufig verkalkte Weichteiltumoranteile und das typische «Codmann Dreieck» als Zeichen einer Periostabhebung zeigt. Eine Kernspintomografie (MRT) des gesamten befallenen Knochens einschliesslich der angrenzenden Gelenke ist in der Regel der nächste Abklärungsschritt. Hiermit können die genaue intramedulläre und extraossäre Ausdehnung sowie die Beziehung zu den neurovaskulären Strukturen am besten dargestellt werden. Weitere Abklärungen beinhalten ein Computertomogramm (CT) der Lungen zum Ausschluss von Lungenmetastasen und eine 99Tc-Knochenszintigrafie zum Ausschluss von Skelettmetastasen. Der Stellenwert einer Positronen-Emissions-Tomografie (PET) ist noch nicht klar (5).
Biopsie Die definitive Diagnose erfolgt stets durch die histologische Aufarbeitung einer repräsentativen Tumorbiopsie. Bei Verdacht auf einen malignen Tumor sollte frühzeitig Kontakt zu einem interdisziplinären Zentrum mit Erfahrung in der Behandlung solcher Tumore erfolgen. Die Biopsien müssen stets sorgfältig im Hinblick auf die definitive Tumorentfernung geplant werden, da der Biopsiekanal unbedingt im späteren Resektionsgebiet liegen muss. Ein unglück-
ONKOLOGIE 4/2009
Im Fokus: Tumoren im Kindesalter
lich gewählter Biopsiekanal kann unter Umständen einen späteren Extremitätenerhalt verunmöglichen (6). Neben der histologischen Aufarbeitung gehören inzwischen immunhistochemische und molekulargenetische Untersuchungen zum Standard.
Behandlungsstrategien Die Behandlung des OS umfasst die komplette Tumorresektion im Gesunden sowie eine Polychemotherapie. Da in historischen Patientenkollektiven mehr als 80% aller Fälle nach alleiniger chirurgischer Behandlung Lungenmetastasen entwickelten, müssen okkulte Metastasen bei den meisten Patienten bereits initial vorhanden sein und begründen die absolute Notwendigkeit einer systemischen Behandlung. Die wirksamsten Substanzen sind hochdosiertes Methotrexat, Doxorubicin, Cisplatin und Ifosfamid. Eine Kombinationstherapie dieser Substanzen ist einer Monotherapie überlegen (7). Die derzeit praktizierte neoadjuvante Chemotherapie mit zirka 2 bis 3 Monaten präoperativer Behandlung bietet die Vorteile eines unverzüglichen Behandlungsbeginns ohne Verzögerung durch Wundheilungsprobleme von grösseren Tumoroperationen. Es entsteht ein Zeitgewinn für die Operationsplanung, und das histologische Ansprechen auf die präoperative Chemotherapie liefert wertvolle prognostische Informationen. Die adjuvante Chemotherapie wird nach der Operation für mindestens 5 bis 6 Monate fortgesetzt. Die Radiotherapie besitzt im interdisziplinären Behandlungskonzept nur einen geringen Stellenwert. Einzige verlässlich wirksame Lokaltherapie ist die komplette Tumorresektion, die dafür spezialisierten Tumororthopäden vorbehalten sein sollte. In den letzten 30 Jahren konnten Amputationen bis auf einen Anteil von 10 bis 20% durch Extremitäten erhaltende Eingriffe ersetzt werden (8). Die Tumorresektion muss stets unter Einhaltung weiter oder radikaler Resektionsgrenzen erfolgen, das heisst, die Entfernung des Tumors erfolgt in einer Schicht gesunden Gewebes.
Prognose Mit dem Einsatz intensiver Chemotherapien hat sich die Prognose in den letzten
30 Jahren entscheidend verbessert. Die Heilungsraten für nichtmetastasierte OS liegen mit der derzeitigen Behandlungsstrategie bei 60 bis 70%, für Patienten mit initialen Metastasen bei unter 30%. Patienten mit gutem Ansprechen auf die präoperative Chemotherapie haben eine deutlich bessere Zehn-Jahres-Überlebenswahrscheinlichkeit als die mit schlechtem Ansprechen (73% vs. 47%; 2). Leider erleiden etwa 25 bis 30% der Patienten ein Rezidiv, dieses tritt meist in den Lungen auf. Eine erneute Aussicht auf Heilung besteht mit chirurgischer Entfernung aller Metastasen, der Stellenwert einer erneuten Chemotherapie wird kontrovers diskutiert (9, 10).
Ewing-Sarkom-Familie
Ewing-Sarkome (EWS) sind eine klinisch und immunhistochemisch heterogene, molekular jedoch einheitliche Gruppe maligner, klein-rund-blauzelliger Tumore. Dazu gehören: ▲ das klassische Ewing-Sarkom, das
gewöhnlich knöchernen Ursprungs ist, ▲ die peripheren malignen primitiven neuroektodermalen Tumoren (PNET) sowie ▲ die Askin-Tumoren der Thoraxwand. Das EWS ist der zweithäufigste Knochentumor bei Kindern und Heranwachsenden; das mediane Alter bei Diagnosestellung ist 15 Jahre. Es sind mehr Knaben betroffen mit einem Geschlechterverhältnis von 1,5:1. Gelegentlich tritt der Tumor in den Weichteilen auf. Es gibt keine Hinweise für eine genetische Prädisposition; interessant ist jedoch das fast völlige Fehlen von EWS in der afrikanischen und chinesischen Population (11). In über 95% der Patienten kann eine Translokation t(11;22) oder eine Variante davon, wie t(21;22), detektiert werden, wobei es zur Genfusion EWS-FLI1 oder EWS-ERG kommt. Es können sowohl die langen Röhrenknochen als auch das Achsenskelett betroffen sein, eine diaphysäre Lage ist typisch. 25% der Patienten haben initial eine nachweisbare Metastasierung in die Lungen oder Knochen (12).
Klinische Präsentation Ähnlich wie beim Osteosarkom (OS) sind Schmerz und Schwellung die häufigsten Symptome. Gelegentlich treten auch Fie-
Abbildung 1a: Osteosarkom der Metaphyse des distalen Femurs, destruierende Knochenläsion mit lytischer und sklerotischer Komponente und verkalktem Weichteiltumor (Pfeil) und Codman-Dreieck (Dreieck).
Abbildung 1b: Ewing-Sarkom des Radius, diaphysär gelegen mit Mottenfrassnekrosen und Spiculae (Dreieck) und Zwiebelschalenmuster (Pfeil) des Periosts.
ber und Gewichtsverlust auf, was zur Differenzialdiagnose einer Osteomyelitis führen kann. Bei einigen Patienten findet sich eine Leukozytose, eine beschleunigte Blutsenkungsreaktion oder eine erhöhte LDH.
ONKOLOGIE 4/2009
15
Im Fokus: Tumoren im Kindesalter
Abbildung 2b: Embryonales Rhabdomyosarkom, von der Blasenwand ausgehend, bei einem dreijährigen Knaben.
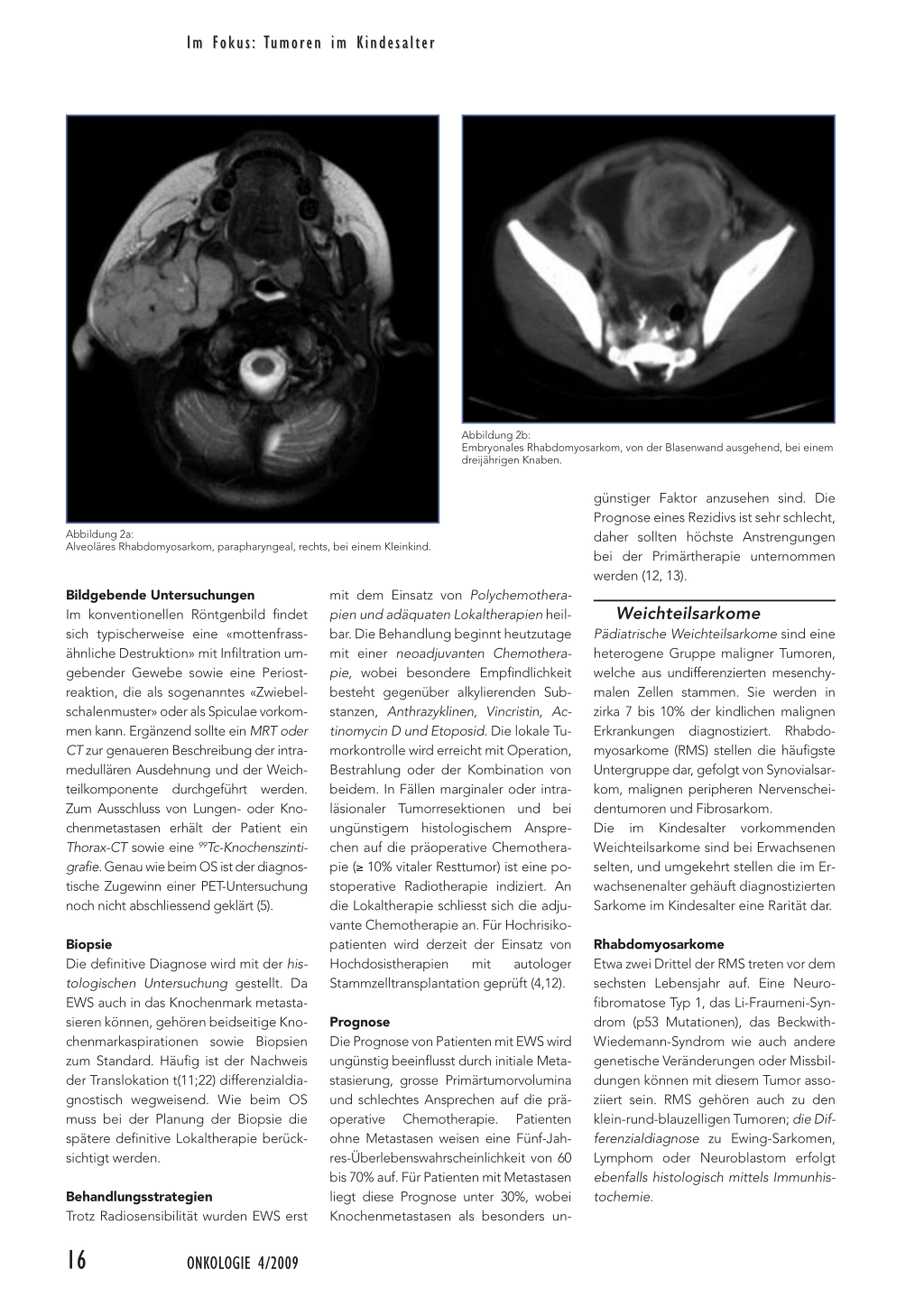
Abbildung 2a: Alveoläres Rhabdomyosarkom, parapharyngeal, rechts, bei einem Kleinkind.
Bildgebende Untersuchungen Im konventionellen Röntgenbild findet sich typischerweise eine «mottenfrassähnliche Destruktion» mit Infiltration umgebender Gewebe sowie eine Periostreaktion, die als sogenanntes «Zwiebelschalenmuster» oder als Spiculae vorkommen kann. Ergänzend sollte ein MRT oder CT zur genaueren Beschreibung der intramedullären Ausdehnung und der Weichteilkomponente durchgeführt werden. Zum Ausschluss von Lungen- oder Knochenmetastasen erhält der Patient ein Thorax-CT sowie eine 99Tc-Knochenszintigrafie. Genau wie beim OS ist der diagnostische Zugewinn einer PET-Untersuchung noch nicht abschliessend geklärt (5).
Biopsie Die definitive Diagnose wird mit der histologischen Untersuchung gestellt. Da EWS auch in das Knochenmark metastasieren können, gehören beidseitige Knochenmarkaspirationen sowie Biopsien zum Standard. Häufig ist der Nachweis der Translokation t(11;22) differenzialdiagnostisch wegweisend. Wie beim OS muss bei der Planung der Biopsie die spätere definitive Lokaltherapie berücksichtigt werden.
Behandlungsstrategien Trotz Radiosensibilität wurden EWS erst
mit dem Einsatz von Polychemotherapien und adäquaten Lokaltherapien heilbar. Die Behandlung beginnt heutzutage mit einer neoadjuvanten Chemotherapie, wobei besondere Empfindlichkeit besteht gegenüber alkylierenden Substanzen, Anthrazyklinen, Vincristin, Actinomycin D und Etoposid. Die lokale Tumorkontrolle wird erreicht mit Operation, Bestrahlung oder der Kombination von beidem. In Fällen marginaler oder intraläsionaler Tumorresektionen und bei ungünstigem histologischem Ansprechen auf die präoperative Chemotherapie (≥ 10% vitaler Resttumor) ist eine postoperative Radiotherapie indiziert. An die Lokaltherapie schliesst sich die adjuvante Chemotherapie an. Für Hochrisikopatienten wird derzeit der Einsatz von Hochdosistherapien mit autologer Stammzelltransplantation geprüft (4,12).
Prognose Die Prognose von Patienten mit EWS wird ungünstig beeinflusst durch initiale Metastasierung, grosse Primärtumorvolumina und schlechtes Ansprechen auf die präoperative Chemotherapie. Patienten ohne Metastasen weisen eine Fünf-Jahres-Überlebenswahrscheinlichkeit von 60 bis 70% auf. Für Patienten mit Metastasen liegt diese Prognose unter 30%, wobei Knochenmetastasen als besonders un-
günstiger Faktor anzusehen sind. Die Prognose eines Rezidivs ist sehr schlecht, daher sollten höchste Anstrengungen bei der Primärtherapie unternommen werden (12, 13).
Weichteilsarkome
Pädiatrische Weichteilsarkome sind eine heterogene Gruppe maligner Tumoren, welche aus undifferenzierten mesenchymalen Zellen stammen. Sie werden in zirka 7 bis 10% der kindlichen malignen Erkrankungen diagnostiziert. Rhabdomyosarkome (RMS) stellen die häufigste Untergruppe dar, gefolgt von Synovialsarkom, malignen peripheren Nervenscheidentumoren und Fibrosarkom. Die im Kindesalter vorkommenden Weichteilsarkome sind bei Erwachsenen selten, und umgekehrt stellen die im Erwachsenenalter gehäuft diagnostizierten Sarkome im Kindesalter eine Rarität dar.
Rhabdomyosarkome Etwa zwei Drittel der RMS treten vor dem sechsten Lebensjahr auf. Eine Neurofibromatose Typ 1, das Li-Fraumeni-Syndrom (p53 Mutationen), das BeckwithWiedemann-Syndrom wie auch andere genetische Veränderungen oder Missbildungen können mit diesem Tumor assoziiert sein. RMS gehören auch zu den klein-rund-blauzelligen Tumoren; die Differenzialdiagnose zu Ewing-Sarkomen, Lymphom oder Neuroblastom erfolgt ebenfalls histologisch mittels Immunhistochemie.
16 ONKOLOGIE 4/2009
Im Fokus: Tumoren im Kindesalter
Klinische Präsentation der RMS RMS können an fast allen Lokalisationen des Körpers auftreten. Gewisse Regionen wie Kopf- und Halsbereich (40%), Urogenitalbereich (20%) und Extremitäten (20%) werden aber bevorzugt. Entsprechend ist die Präsentation von der Primärlokalisation bestimmt. Orbitale RMS zeigen meist eine Verdrängung des Bulbus, nasopharyngeale RMS können durch Schwellung, Nasenhöhlenobstruktion oder polypartige Wucherungen erkenntlich werden. Eine ungünstige prognostische Bedeutung kommt den parameningealen Lokalisationen zu, die den Befall der Meningen respektive eine intrakranielle Ausdehnung beinhalten. Tumoren im Urogenitalbereich können bei Blasen- oder Prostatalokalisation typischerweise zur Obstruktion des Harnabflusses oder Blutung führen. Bei Befall der Vagina oder des Uterus können wiederum polypartige Veränderungen im Vordergrund stehen (14). RMS der Extremitäten präsentieren sich oft mit einer indolenten derben, rasch wachsenden Schwellung.
Bildgebende Untersuchungen Wie bei anderen Sarkomen sind Ultraschall sowie MRT oder allenfalls CT die üblichen bildgebenden Methoden, um die genaue Lokalisation und Ausdehnung des Tumors festzuhalten. Zur Prüfung von Lungenmetastasen wird neben dem Thoraxröntgenbild ein Thorax-CT gefordert. Eine Knochenszintigrafie wird mehrheitlich empfohlen. Je nach Lokalisation des Primärtumors kommt der radiologischen Überprüfung regionaler Lymphknoten (z.B. Beckenlymphknoten bei RMS der unteren Extremität) eine besondere Bedeutung zu. Das genaue Staging ist für die spätere Behandlungsstrategie essenziell. Die Bedeutung der PET-Untersuchung bei RMS ist immer noch kontrovers und ist üblicherweise nicht routinemässig vorgesehen. Die Spezifität des PET kann aber bis zu 95% erreichen (15).
Biopsie Eine Biopsie ist für die Diagnosesicherung und Subklassifizierung absolut notwendig. Eine präzise Diagnostik ist nicht immer ganz einfach, aber entscheidend für die Prognose und damit die weitere
Behandlungsstrategie. Für die Klassifizierung sind Immunhistochemie und heutzutage auch molekulargenetische Untersuchungen unverzichtbar. Bevorzugt wird deshalb eine offene Biopsie, auch wenn mit einer Nadelbiopsie eine Diagnose gestellt werden kann. Üblicherweise wird auch eine Knochenmarkbiopsie im Rahmen des Tumorstagings gefordert. Es gibt vor allem zwei histologische Subtypen, das embryonale RMS und das alveoläre RMS. Seltenere Varianten sind das spindelzellige, das botryoide oder das pleomorphe RMS. Das prognostisch günstigere embryonale RMS wird in etwa 80% der pädiatrischen RMS gefunden, das prognostisch ungünstigere alveoläre RMS, welches durch eine typische Translokation t(2;13) oder, seltener, t(1;13) charakterisiert ist, tritt vermehrt auch an den Extremitäten, vor allem im Adoleszentenalter auf. Die resultierenden Genfusionen PAX3-FKHR beziehungsweise PAX7FKHR resultieren in einer Alteration der biologischen Aktivität von Proteinen, welche Tumorzellwachstum, Apoptose, Differenzierung und Zellmortalität beeinflussen (16).
Behandlungsoptionen bei RMS Die Behandlung von Rhabdomyosarkomen verlangt einen multidisziplinären Ansatz. Im Rahmen interdisziplinärer Diskussionen mit pädiatrischen Onkologen, Chirurgen, Strahlentherapeuten, Pathologen und Radiologen wird die für den Patienten individuell beste Behandlungsstrategie erarbeitet. Eine Chemotherapie basierend auf Ifosfamid, Vincristin, Actinomycin-D und gelegentlich Anthrazyklinen ist Standard. Je nach Tumoransprechen, Histologie, Tumorlokalisation und Tumorgrösse werden zusätzliche Therapiemodalitäten wie Tumorresektion und oder Strahlentherapie angewendet (17). Die primäre Tumorexzision ist nur in denjenigen Fällen anzustreben, bei denen mit Sicherheit der ganze Tumor im Gesunden entfernt werden kann, ohne eine Mutilierung hervorzurufen. Strahlentherapeutische Verfahren beinhalten die konventionelle Photonenbestrahlung oder bei besonderen Lokalisationen die Protonentherapie, die aber europaweit nur in ganz wenigen Zentren
zur Verfügung steht. Damit kann allenfalls – bei gleicher Strahlenwirkung auf den Tumor – gesundes Gewebe besser geschont werden. Diese Protonen-Strahlenanwendung spielt natürlich besonders bei kleinen Kindern eine wichtige Rolle, um Spätfolgen der Strahlentherapie einzuschränken. Auch die Brachytherapie bleibt spezialisierten Zentren vorbehalten und kann in individuellen Situationen angezeigt sein.
Prognose
Die Überlebensrate bei Rhabdomyosar-
komen ist zwischen 1970 und 1995 von
25% bis heute auf 75% gestiegen (18).
Dies ist sicherlich zu einem wesentlichen
Teil den randomisierten klinischen Stu-
dien zu verdanken. Mitentscheidend
sind frühe Diagnose und Überweisung
eines betroffenen Kindes in ein speziali-
siertes Zentrum.
Die Prognose ist vor allem abhängig vom
histologischen Subtyp, von der Tumor-
lokalisation und Ausdehnung und allen-
falls vom Alter des Patienten. Die Hei-
lungschancen von metastatischen Sarko-
men ist auch mit den heutigen
Therapiemöglichkeiten dagegen immer
noch bescheiden (19).
Langfristige Nebenwirkungen sind je
nach Tumorlokalisation und angewende-
ten Therapiemodalitäten teils nicht unbe-
trächtlich. Es gilt deshalb heutzutage, die
Balance zwischen therapeutischer Ag-
gressivität und damit optimalen Hei-
lungschancen gegenüber einer eher mo-
deraten Therapie mit potenziell höherem
Rezidivrisiko, aber weniger langfristigen
Therapiefolgen zu finden.
▲
Prof. Dr. med. Felix K. Niggli (Korrespondenzadresse) Abteilungsleiter Pädiatrische Onkologie Universitäts-Kinderkliniken Zürich 8032 Zürich E-Mail: felix.niggli@kispi.uzh.ch
sowie Dr. med. Nicole Bodmer Abteilung Pädiatrische Onkologie Universitäts-Kinderkliniken Zürich 8032 Zürich
ONKOLOGIE 4/2009
17
Im Fokus: Tumoren im Kindesalter
Merksätze
▲ Sarkome sind teilweise durch typische Chromosomentranslokationen charakterisiert, die vor allem diagnostische, aber auch prognostische Bedeutung haben.
▲ Chemotherapie ist Bestandteil aller Behandlungen der genannten kindlichen Sarkome. ▲ Ein spezialisiertes, interdisziplinäres Behandlungsteam soll bereits bei Verdacht auf ein kindliches
Sarkom konsultiert werden, da (z.B.) bereits die Biopsie mitentscheidend sein kann für das langfristige Resultat der Behandlung. ▲ Heilungschancen bei Sarkomen haben sich mit einer multimodalen Therapie im Rahmen von randomisierten, internationalen Studien in den vergangenen 30 Jahren kontinuierlich verbessert.
Quellen:
1. Fuchs B, Pritchard DJ.: Etiology of osteosarcoma. Clin Orthop Relat Res 2002(397): 40–52.
2. Bielack SS, Kempf-Bielack B, et al.: Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 2002; 20(3): 776–90.
3. Bacci G, Ferrari S, et al.: Nonmetastatic osteosarcoma of the extremity with pathologic fracture at presentation: local and systemic control by amputation or limb salvage after preoperative chemotherapy. Acta Orthop Scand 2003; 74(4): 449–54.
4. Heare T, Hensley MA, Dell’Orfano S.: Bone tumors: osteosarcoma and Ewing’s sarcoma. Curr Opin Pediatr 2009; 21(3): 365–72.
5. Meyer JS, Nadel HR, et al.: Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children’s Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 2008; 51(2): 163–70.
6. Heare TC, Enneking WF, Heare MM.: Staging techniques and biopsy of bone tumors. Orthop Clin North Am 1989; 20(3): 273–85.
7. Link MP, Goorin AM, et al.: Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the Multi-Institutional Osteosarcoma Study. Clin Orthop Relat Res 1991(270): 8–14.
8. Sluga M, Windhager R, et al.: Local and systemic control after ablative and limb sparing surgery in patients with osteosarcoma. Clin Orthop Relat Res 1999(358): 120–27.
9. Kempf-Bielack B, et al.: Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol 2005; 23(3): 559–68.
10. Ferrari S, Briccoli A, et al.: Postrelapse survival in osteosarcoma of the extremities: prognostic factors for long-term survival. J Clin Oncol 2003; 21(4): 710–15.
11. Hense HW, Ahrens S, et al.: Descriptive epidemiology of Ewing’s tumor – analysis of German patients from (EI)CESS 1980-1997]. Klin Pädiatr 1999; 211(4): 271–75.
12. Paulussen M, Frohlich B, Jurgens H.: Ewing tumour: incidence, prognosis and treatment options. Paediatr Drugs 2001; 3(12): 899–913.
13. Caudill JS, Arndt CA.: Diagnosis and management of bone malignancy in adolescence. Adolesc Med State Art Rev 2007; 18(1): 62–78.
14. Herzog CE, Stewart JM, Blakely ML.: Pediatric soft tissue sarcomas. Surg Oncol Clin N Am 2003; 12(2): 419–47.
15. Klem ML, Grewal RK, et al.: PET for staging in rhabdomyosarcoma: an evaluation of PET as an adjunct to current staging tools. J Pediatr Hematol Oncol 2007; 29(1): 9–14.
16. Barr FG.: Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001; 20(40): 5736–46.
17. Stevens MC, Rey A, et al.: Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology—SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol 2005; 23(12): 2618–28.
18. Pappo AS, Shapiro DN, et al.: Biology and therapy of pediatric rhabdomyosarcoma. J Clin Oncol 1995; 13(8): 2123–39.
19. Oberlin O, Rey A, et al.: Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol 2008; 26(14): 2384–9.
18 ONKOLOGIE 4/2009