
Transkript
Neue Therapien
Neuentwicklung und Marktzulassung
Erstes Biosimilar zu Filgrastim
Ablaufende Patentschutzfristen biotechnologisch hergestellter Arzneimittel ermögli- der Phasen I und III nachgewiesen werchen seit Kurzem die Produktion preisgünstiger Nachahmerprodukte. Der granulo- den.
zytenkolonienstimulierende Faktor Filgrastim-Mepha® ist das erste Biosimilar zu Neupogen® in der Schweiz. Das Medikament wird hauptsächlich im Rahmen von
Chemotherapien zur Verringerung der Dauer, Häufigkeit und Schwere von Neutro-
Filgrastim von Mepha
Filgrastim-Mepha® ist das erste Biosimilar zu Neupogen® und voraussichtlich
das erste in der Schweiz zugelassene Bio-
penien angewendet.
similar. Bei dem Präparat handelt es sich um einen granulozytenkolonienstimulie-
renden Faktor (G-CSF), der aus 175 Aufgrund ihrer komplexen Strukturen und Weil die Herstellungsart bei Biopharma- Aminosäuren besteht. Filgrastim-Mepha®
der Herstellung in lebenden Zellen sind zeutika eine entscheidende Rolle spielt, wird in Escherichia-coli-Bakterien gewon-
Biopharmazeutika einzigartig und nicht bezieht sich der Patentschutz auf diesen nen, in die zuvor eine synthetisch herge-
vollständig kopierbar. Somit handelt es Prozess. Die Moleküle selbst, wie Insulin stellte Kopie des humanen G-CSF-Gens
sich bei den Nachfolgepräparaten nicht oder Wachstumsfaktoren, sind nicht pa- eingefügt wurde. Das Medikament för-
um Biogenerika, sondern um strukturähn- tentgeschützt, da es sich um körpereige- dert die Bildung von neutrophilen Gra-
liche Biosimilars. Für die Marktzulassung ne Stoffe handelt.
nulozyten und wirkt Neutropenien ent-
muss deren therapeutische Äquivalenz Biosimilars unterscheiden sich von che- gegen, die als wesentlicher Risikofaktor
mit dem Referenzprodukt des Erstanbie- misch definierten, synthetisch hergestell- infektionsbedingter Morbidität und Mor-
ters anhand präklinischer und klinischer ten Wirkstoffen in verschiedener Hinsicht talität gelten.
Studien nachgewiesen werden.
(vgl. Tabelle).
Von der Therapie profitieren vor allem
Biosimilars anders als Generika
Zulassungsverfahren für Biosimilars
Krebspatienten, die sich einer Chemotherapie unterziehen. Filgrastim reguliert im Knochenmark die Bildung und Diffe-
Biosimilars sind Nachfolgeprodukte bio- Da ein Generikum immer die gleiche renzierung der neutrophilen Granulo-
technologisch hergestellter Referenz- Wirksubstanz enthält wie das Original, zyten sowie deren Freisetzung ins Blut.
präparate, deren Patentschutzfrist abge- reichen für die Zulassung meist Bioäqui- Klinisch macht man sich diesen Effekt zu-
laufen ist. Ihre Wirkung, Verträglichkeit valenzstudien aus, in denen bei einer li- nutze, indem man mit Filgrastim die Re-
und Sicherheit ist vergleichbar mit den Re- mitierten Anzahl gesunder Probanden generation der Granulozyten nach einer
ferenzprodukten, ihre Struktur kann sich vergleichbare Plasmaspiegelkurven be- Zytostatikatherapie beschleunigt. Unter
jedoch von der Referenzsubstanz unter- legt werden. Die Marktzulassung für Bio- der Therapie produziert das Knochen-
scheiden. Aufgrund ihrer Komplexität und similars ist dagegen um einiges aufwän- mark innerhalb kurzer Zeit (4 bis 5 Stun-
der Herstellung in lebenden Zellen sind diger. Aufgrund der biologischen Variabi- den) neutrophile Granulozyten und gibt
Biopharmazeutika einzigartig und, im Ge- lität muss die therapeutische Äquivalenz diese ins Blut ab. Ohne das Medikament
gensatz zu chemisch synthetisierten Wirk- mit dem Referenzprodukt anhand präkli- würde dieser Prozess etwa fünf Tage dau-
stoffen, nicht vollständig kopierbar.
nischer Studien sowie klinischer Studien ern. Mit Filgrastim lassen sich somit Dau-
Während es sich bei chemisch syntheti-
sierten Substanzen wie Acetylsalicylsäure um kleine Moleküle handelt, sind biotechnologisch hergestellte Wirkstoffe meist 100- bis 1000-fach grösser. Auf-
Tabelle: Unterschied strukturidentische Generika – strukturähnliche Biosimilars
grund der komplexen räumlichen Strukturen kann bei den Proteinen eine Variabilität auftreten, beispielsweise in der Tertiärstruktur oder beim Glykosilierungsmuster. Diese Unterschiede können die Wirksamkeit des Produkts beeinflussen und werden unter anderem durch die Bedingungen des Herstellungsprozesses oder den Umgang mit der Sub-
Chemisch synthetisierte Arzneimittel Nachfolger: strukturidentische Generika chemische Synthese, einfaches Herstellungsverfahren klar definierte Substanz mit einfacher Struktur feste Bindung zwischen einzelnen Atomen geringes Molekulargewicht chemisch stabile Wirkstoffe Molekül ist patentgeschützt Immunreaktionen selten Wechselwirkungen mit anderen Medikamenten häufig
Biotechnologisch hergestellte Arzneimittel Nachfolger: strukturähnliche Biosimilars komplexer Herstellungsprozess in lebenden Zellen Protein mit hochkomplexer Struktur Molekül mit stabilen und instabilen Verbindungen sehr hohes Molekulargewicht empfindlich gegenüber physikalischen Einflüssen Herstellungsprozess ist patentgeschützt Immunreaktionen möglich Wechselwirkungen mit anderen Medikamenten selten
stanz mitbestimmt.
ONKOLOGIE 1/2009
31
Neue Therapien
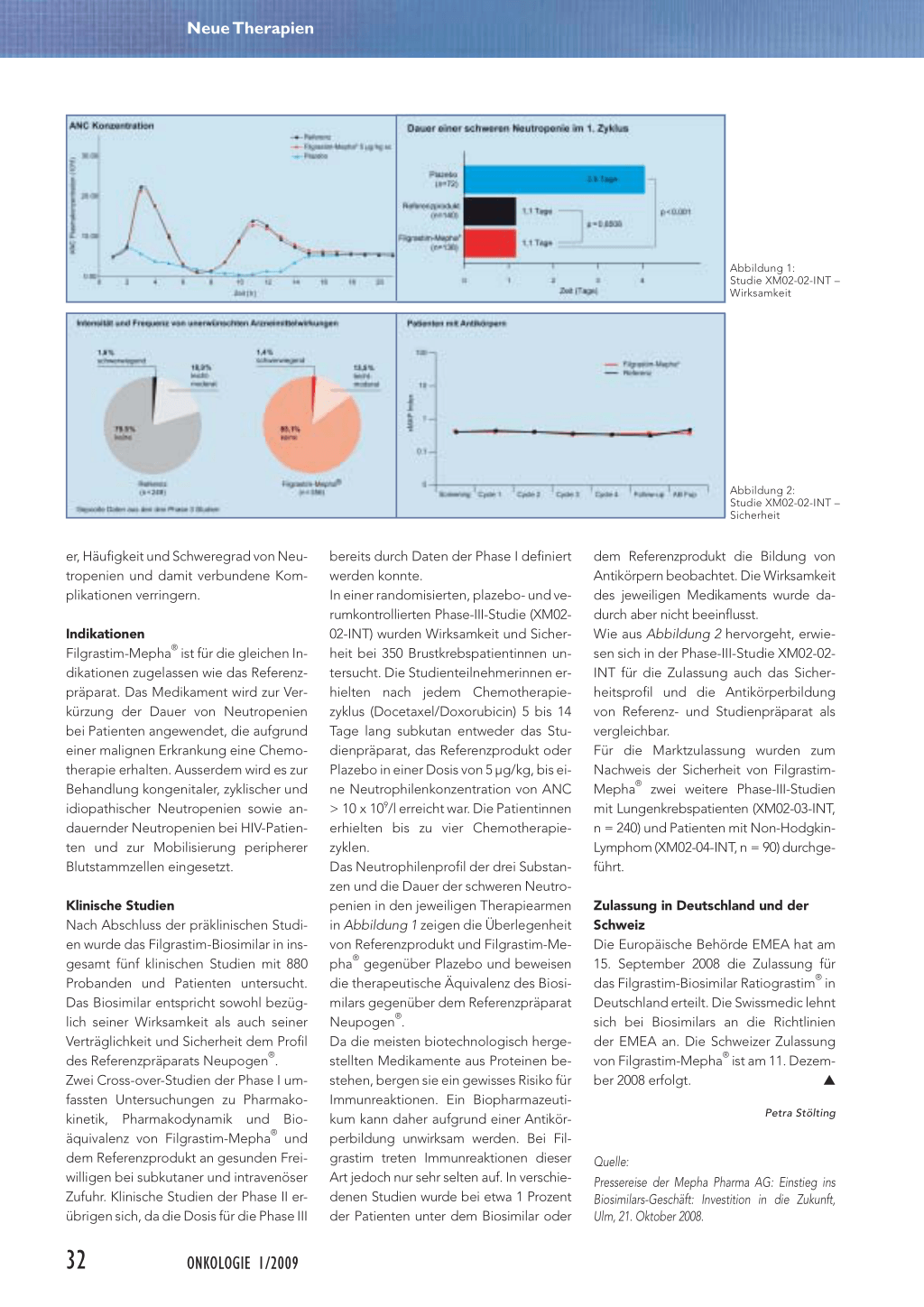
Abbildung 1: Studie XM02-02-INT – Wirksamkeit
Abbildung 2: Studie XM02-02-INT – Sicherheit
er, Häufigkeit und Schweregrad von Neutropenien und damit verbundene Komplikationen verringern.
Indikationen Filgrastim-Mepha® ist für die gleichen Indikationen zugelassen wie das Referenzpräparat. Das Medikament wird zur Verkürzung der Dauer von Neutropenien bei Patienten angewendet, die aufgrund einer malignen Erkrankung eine Chemotherapie erhalten. Ausserdem wird es zur Behandlung kongenitaler, zyklischer und idiopathischer Neutropenien sowie andauernder Neutropenien bei HIV-Patienten und zur Mobilisierung peripherer Blutstammzellen eingesetzt.
Klinische Studien Nach Abschluss der präklinischen Studien wurde das Filgrastim-Biosimilar in insgesamt fünf klinischen Studien mit 880 Probanden und Patienten untersucht. Das Biosimilar entspricht sowohl bezüglich seiner Wirksamkeit als auch seiner Verträglichkeit und Sicherheit dem Profil des Referenzpräparats Neupogen®. Zwei Cross-over-Studien der Phase I umfassten Untersuchungen zu Pharmakokinetik, Pharmakodynamik und Bioäquivalenz von Filgrastim-Mepha® und dem Referenzprodukt an gesunden Freiwilligen bei subkutaner und intravenöser Zufuhr. Klinische Studien der Phase II erübrigen sich, da die Dosis für die Phase III
bereits durch Daten der Phase I definiert werden konnte. In einer randomisierten, plazebo- und verumkontrollierten Phase-III-Studie (XM0202-INT) wurden Wirksamkeit und Sicherheit bei 350 Brustkrebspatientinnen untersucht. Die Studienteilnehmerinnen erhielten nach jedem Chemotherapiezyklus (Docetaxel/Doxorubicin) 5 bis 14 Tage lang subkutan entweder das Studienpräparat, das Referenzprodukt oder Plazebo in einer Dosis von 5 µg/kg, bis eine Neutrophilenkonzentration von ANC > 10 x 109/l erreicht war. Die Patientinnen erhielten bis zu vier Chemotherapiezyklen. Das Neutrophilenprofil der drei Substanzen und die Dauer der schweren Neutropenien in den jeweiligen Therapiearmen in Abbildung 1 zeigen die Überlegenheit von Referenzprodukt und Filgrastim-Mepha® gegenüber Plazebo und beweisen die therapeutische Äquivalenz des Biosimilars gegenüber dem Referenzpräparat Neupogen®. Da die meisten biotechnologisch hergestellten Medikamente aus Proteinen bestehen, bergen sie ein gewisses Risiko für Immunreaktionen. Ein Biopharmazeutikum kann daher aufgrund einer Antikörperbildung unwirksam werden. Bei Filgrastim treten Immunreaktionen dieser Art jedoch nur sehr selten auf. In verschiedenen Studien wurde bei etwa 1 Prozent der Patienten unter dem Biosimilar oder
dem Referenzprodukt die Bildung von Antikörpern beobachtet. Die Wirksamkeit des jeweiligen Medikaments wurde dadurch aber nicht beeinflusst. Wie aus Abbildung 2 hervorgeht, erwiesen sich in der Phase-III-Studie XM02-02INT für die Zulassung auch das Sicherheitsprofil und die Antikörperbildung von Referenz- und Studienpräparat als vergleichbar. Für die Marktzulassung wurden zum Nachweis der Sicherheit von FilgrastimMepha® zwei weitere Phase-III-Studien mit Lungenkrebspatienten (XM02-03-INT, n = 240) und Patienten mit Non-HodgkinLymphom (XM02-04-INT, n = 90) durchgeführt.
Zulassung in Deutschland und der
Schweiz
Die Europäische Behörde EMEA hat am
15. September 2008 die Zulassung für das Filgrastim-Biosimilar Ratiograstim® in
Deutschland erteilt. Die Swissmedic lehnt
sich bei Biosimilars an die Richtlinien
der EMEA an. Die Schweizer Zulassung von Filgrastim-Mepha® ist am 11. Dezem-
ber 2008 erfolgt.
▲
Petra Stölting
Quelle:
Pressereise der Mepha Pharma AG: Einstieg ins Biosimilars-Geschäft: Investition in die Zukunft, Ulm, 21. Oktober 2008.
32 ONKOLOGIE 1/2009