



Transkript
Im Fokus: Maligne Lymphome und multiples Myelom
Follikuläre Lymphome
Inzidenz, Pathogenese, Diagnostik,Therapiestrategien Follikuläre Lymphome bilden die zweithäufigste Gruppe der Non-Hodgkin-Lymphome. Im Stadium I und II kann die Bestrahlung eine kurative Chance bieten, während die Heilung bei den fortgeschrittenen Stadien noch unerreicht ist. Rituximab hat seinen festen Platz in der Erstlinien- wie auch in der Erhaltungstherapie. Die Radioimmunbehandlung gewinnt zunehmend an Bedeutung. Der Stellenwert der konventionellen Zytostatika wird in Zukunft neu zu definieren sein.
WALTER MINGRONE, ALDEN MOCCIA
Die follikulären Lymphome (FL) bilden die zweithäufigste Gruppe der Non-Hodgkin-Lymphome (NHL). Innerhalb der lymphoproliferativen Malignome stellen die FL nach den diffus grosszelligen NHL, den Plasmazellerkrankungen und der CLL/SLL die vierthäufigste Gruppe dar. Die Inzidenz beträgt in der weissen Bevölkerung der westlichen Welt rund 3,8/100 000, ist weiterhin steigend und fällt gegenüber den asiatischen und afroamerikanischen Einwohnern nahezu doppelt so hoch aus (1–3). Im Gegensatz zu anderen Formen der NHL konnte bei den FL keine Assoziation mit kongenitalen oder erworbenen Formen der Immundefizienz, mit Atopien oder Autoimmunerkrankungen nachgewiesen werden. Unklar bleibt, ob der Gebrauch von Haarfärbemitteln das Auftreten von FL begünstigt (4). Die Anamnese eines an FL erkrankten Familienangehörigen erhöht das prädisponierende Risiko um den Faktor 2,6 (5).
Pathogenese, Histopathologie und Mikroinvironment
Das FL entstammt den B-Zellen des Keimzentrums in den Follikeln der Lymphknoten. In 80 bis 90% der Fälle ist es durch die Translokation t(14; 18) charakterisiert, die letztlich dazu führt, dass das anti-apoptotisch wirkende bcl-2 Protein überexprimiert wird (6). Die morphologische Einteilung der FL in die Grade 1 bis 3 richtet sich nach der durchschnittlichen Zahl der Zentroblasten in zehn untersuchten neoplastischen Follikeln bei 40-facher Vergrösserung (high power microscopic field [HPF]) und hat prognostische Bedeutung (7) (Tabelle 1). So weisen FL Grad 3 ein aggressives Verhalten auf, sie werden in der Regel analog den diffus grosszelligen NHL behandelt.
Die histologische Diagnostik erfordert eine Lymphknotenbiopsie. Eine übereinstimmende Gradierung durch verschiedene Pathologen ist allein aufgrund der Histomorphologie schwierig zu erzielen. Die Immunhistochemie kann zur eindeutigeren Klassifizierung beitragen (8). Zunehmende Beachtung gewinnt das sogenannte Mikroinvironment, das heisst das Gewebe rund um die malignen Zellen. So konnte in retrospektiven Studien gezeigt werden, dass eine hohe Zahl CD8+-TZellen oder Foxp3-exprimierende T-Zellen (Tregcells, regulatorische T-Zellen) mit einer besseren Prognose einhergehen (9, 10). Anderseits vermochte eine grosse Untersuchung an 289 Patienten keinen bedeutsamen Einfluss der T-Zellen auf das Outcome der FL nachzuweisen (11). Es konnte auch gezeigt werden, dass Rituximab die prognostisch ungünstige Infiltration durch Makrophagen wieder auszugleichen vermag (12). Möglicherweise liegt im Mikroinvironment der Schlüssel zum Verständnis des Phänomens, dass bei FL gelegentlich spontane Remissionen beobachtet werden. So bleibt die Abschätzung der Prognose weiterhin durch die einfachen und gut reproduzierbaren klinischen Parameter gemäss dem Follicular Lymphoma International Prognostic Index (FLIPI-Index) bestimmt. Im Verlauf der Erkrankung ist die Transformation des FL in ein diffus grosszelliges Lymphom nicht selten; dies tritt im Lauf der Erkrankung bei 40 bis 60% aller Patienten ein (13).
Klinische Diagnostik und Staging
Das Krankheitsbild manifestiert sich typischerweise
24 ONKOLOGIE 4/2008
Im Fokus: Maligne Lymphome und multiples Myelom
Tabelle 1:
Morphologische Einteilung der follikulären Lymphome
Entsprechend der durchschnittlichen Zahl der Zentroblasten in 10 untersuchten neoplastischen Follikeln
Grad 1: Grad 2: Grad 3: Grad 3 a:
Grad 3 b:
0 bis 5 Zentroblasten/HPF 6 bis 15 Zentroblasten/HPF > 15 Zentroblasten/HPF gemischt Zentrozyten und Zentroblasten ausschliesslich Zentroblasten
HPF = high power microscopic field
mit einer generalisierten und palpatorisch indolenten Lymphadenopathie. Zu Beginn der Erkrankung ist der Allgemeinzustand in den meisten Fällen gut, und nur in einer Minderheit werden B-Symptome beklagt. Die Festlegung des Stadiums nach Ann Arbor umfasst die Abklärung des Stagings mittels Computertomografie von Hals bis Becken und die Knochenmarkuntersuchung. Im Labor werden neben dem Hämatogramm die Leber- und Nierenfunktionswerte, die LDH, das -2Mikroglobulin und Virusserologien für Hepatitis B und C, EBV, CMV und HIV bestimmt. Ausserhalb von klinischen Studien wird die PET-Untersuchung nicht empfohlen (14). Die grosse Mehrheit (~80%) hat nach Ann Arbor ein Stadium III oder IV. Die Milz ist selten, das Knochenmark in rund 50% der Fälle befallen. Das Alter, das Tumorstadium nach Ann Arbor, der Gehalt an Hämoglobin, die LDH und die Anzahl der befallenen Lymphknotenareale erlauben anhand des FLIPI-Scores, die Prognose des Patienten abzuschätzen (15) (Tabelle 2). Die Aussagekraft des FLIPIScores bleibt auch bei einer Immunochemotherapie wie R-CHOP erhalten (16).
Gemäss FLIPI sind die prognostisch negativen Faktoren: ▲ Alter > 60 Jahre ▲ Ann-Arbor-Stadium III oder IV ▲ LDH > UNL ▲ Befallene Lymphknotenareale > 4 ▲ Hämoglobingehalt < 120 g/l.
Allgemeine Therapiestrategien
Frühstadien Für die Minderheit (10–15%) der Patienten mit FL, die sich bei Diagnose im Stadium I oder II befinden, ist die Bestrahlung («involved field») in kurativer Absicht die Therapie der Wahl (17). Eine retrospektive Analyse von Patienten aus Stanford, die im Frühstadium der Erkrankung keine Behandlung erhielten, ergab bezüglich Gesamtüberlebens vergleichbare Resultate wie bei behandelten Patienten (18). Im Einzelfall kann somit die Strategie des «watchful waiting» erwogen werden.
Fortgeschrittene Stadien mit histologischem Grad 1 und 2 Patienten in fortgeschrittenem Stadium können auch heute (noch) nicht geheilt werden. Die Indikation zur Therapie und die Wahl der individuell am besten geeigneten Behandlung richtet sich nach dem Risiko gemäss dem FLIPI-Score, dem Allgemeinzustand, den Begleiterkrankungen und nicht zuletzt nach den Wünschen des Patienten. Patienten mit tiefem und mittlerem Score haben eine gute Langzeitprognose, weshalb bei asymptomatischen Patienten mit der Aufnahme einer Behandlung zugewartet werden kann. Patienten mit höherem FLIPI-Score haben ein medianes Überleben von rund 50% nach 5 Jahren, und nur etwas mehr als ein Drittel der Patienten erreicht 10 Jahre. In einer solchen Situation ist auch bei symptomfreien Patienten mit dem ra-
Tabelle 2:
Prognose (Gesamtüberleben) entsprechend FLIPI-Score
FLIPI-Score 0–1 2 3–5
Fünf-Jahres-Gesamtüberleben 91% 78% 53%
Zehn-Jahres-Gesamtüberleben 71% 51% 36%
schen Auftreten von Beschwerden zu rechnen. Bei diesen Patienten zielt die Behandlung darauf, sowohl die beschwerdefreie Zeit als auch die gesamte Lebenszeit zu verlängern. In der Regel wird hierfür heute eine Polychemotherapie, kombiniert mit Rituximab, gewählt. Die initiale Monotherapie mit Rituximab, also ohne vorgängige Chemotherapie, ist in der Schweiz (noch) nicht kassenpflichtig. Die Monochemotherapie mit Chlorambucil oder Fludarabin als Erstlinientherapie wird heute kaum mehr praktiziert, kann aber im Einzelfall eine verhältnismässige Option sein.
Follikuläre Lymphome mit histologischem Grad 3 FL mit einer hohen Zahl an Zentroblasten (>15/HPF) haben eine deutlich schlechtere Prognose. Sie werden analog zu den diffus grosszelligen NHL mit Aussicht auf eine langdauernde Remission und Heilung behandelt (19, 20).
Therapiemodalitäten
Klassische Strahlentherapie Das FL zählt zu den radiosensibelsten Neoplasien. Insbesondere in den Frühstadien können mit einer Gesamtdosis von 36 bis 40 Gy und mit Involved-fieldBestrahlungen lang anhaltende Remissionen erzielt werden (21). Tritt nach einer vorgängigen systemischen Behandlung ein lokal begrenztes Rezidiv auf, kann dieses mit einer tief dosierten Bestrahlung mit 4 Gy erstaunlich gut kontrolliert werden (komplette Remission in rund 60%; mediane Zeit bis zur lokalen Progression von 42 Monaten bei Patienten in lokaler CR) (22).
Radioimmunotherapie (RIT) Die systemische Applikation eines gegen die Tumorzelle gerichteten Antikörpers in Konjugation mit einem Radioisotop erlaubt die gerichtete zytotoxische Bestrahlung der Antigen-tragenden Zellen wie auch der unmittelbaren Umgebung (Cross-fire-Effekt). Zurzeit sind zwei solche gegen die CD20positiven Lymphozyten gerichtete Substanzen auf dem Markt. Das Tositumomab ist mit I131 (Bexxar®) und das Ibritumomab Tiuxetan mit Yttrium90 (Zevalin®) gebunden. Die Wirksamkeit dieser Behandlungen wurde zunächst beim
ONKOLOGIE 4/2008
25
Im Fokus: Maligne Lymphome und multiples Myelom
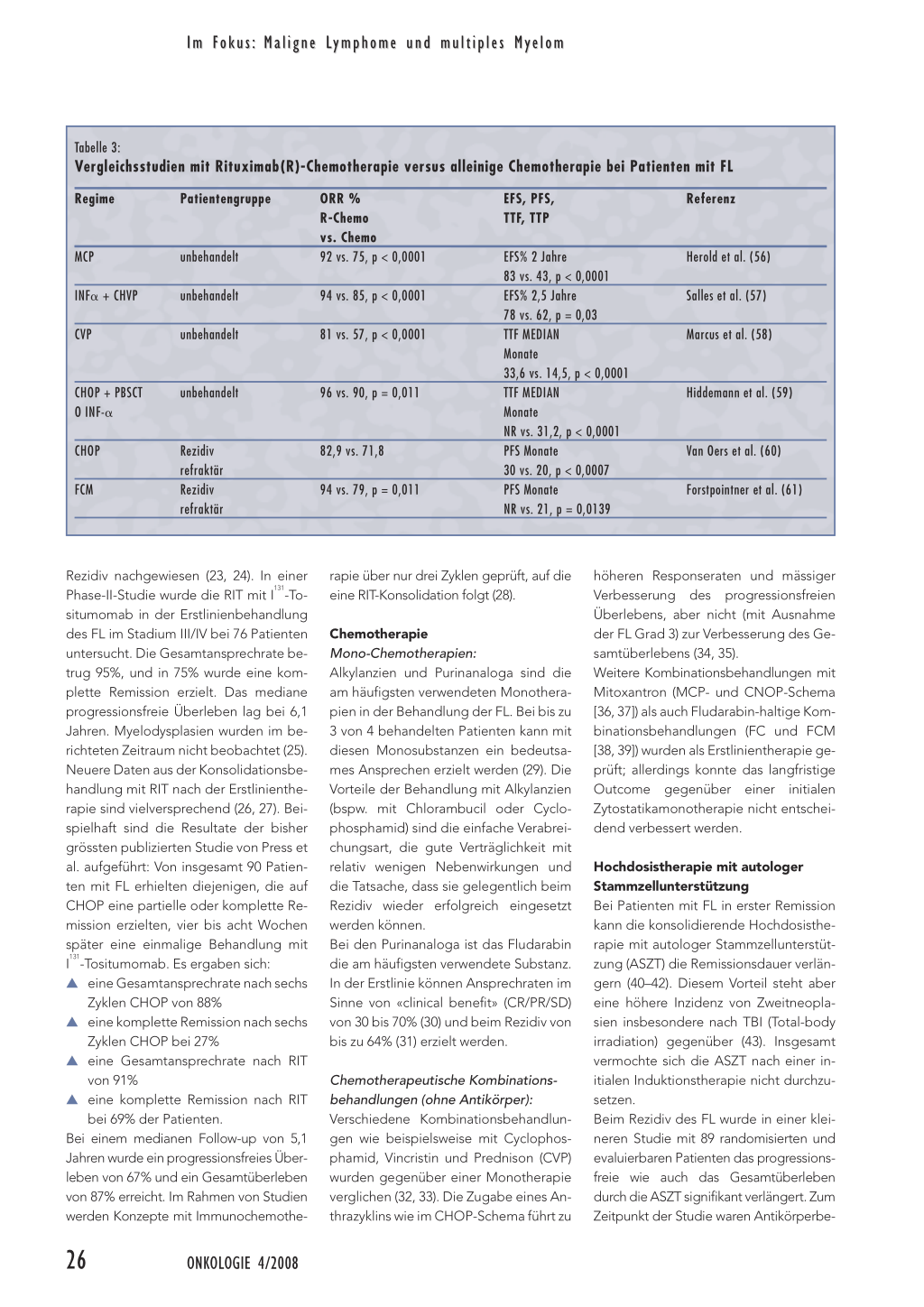
Tabelle 3:
Vergleichsstudien mit Rituximab(R)-Chemotherapie versus alleinige Chemotherapie bei Patienten mit FL
Regime
Patientengruppe
MCP INF␣ + CHVP CVP
unbehandelt unbehandelt unbehandelt
ORR % R-Chemo vs. Chemo 92 vs. 75, p < 0,0001 94 vs. 85, p < 0,0001 81 vs. 57, p < 0,0001 CHOP + PBSCT O INF-␣ CHOP FCM unbehandelt Rezidiv refraktär Rezidiv refraktär 96 vs. 90, p = 0,011 82,9 vs. 71,8 94 vs. 79, p = 0,011 EFS, PFS, TTF, TTP EFS% 2 Jahre 83 vs. 43, p < 0,0001 EFS% 2,5 Jahre 78 vs. 62, p = 0,03 TTF MEDIAN Monate 33,6 vs. 14,5, p < 0,0001 TTF MEDIAN Monate NR vs. 31,2, p < 0,0001 PFS Monate 30 vs. 20, p < 0,0007 PFS Monate NR vs. 21, p = 0,0139 Referenz Herold et al. (56) Salles et al. (57) Marcus et al. (58) Hiddemann et al. (59) Van Oers et al. (60) Forstpointner et al. (61) Rezidiv nachgewiesen (23, 24). In einer Phase-II-Studie wurde die RIT mit I131-Tositumomab in der Erstlinienbehandlung des FL im Stadium III/IV bei 76 Patienten untersucht. Die Gesamtansprechrate betrug 95%, und in 75% wurde eine komplette Remission erzielt. Das mediane progressionsfreie Überleben lag bei 6,1 Jahren. Myelodysplasien wurden im berichteten Zeitraum nicht beobachtet (25). Neuere Daten aus der Konsolidationsbehandlung mit RIT nach der Erstlinientherapie sind vielversprechend (26, 27). Beispielhaft sind die Resultate der bisher grössten publizierten Studie von Press et al. aufgeführt: Von insgesamt 90 Patienten mit FL erhielten diejenigen, die auf CHOP eine partielle oder komplette Remission erzielten, vier bis acht Wochen später eine einmalige Behandlung mit I131-Tositumomab. Es ergaben sich: ▲ eine Gesamtansprechrate nach sechs Zyklen CHOP von 88% ▲ eine komplette Remission nach sechs Zyklen CHOP bei 27% ▲ eine Gesamtansprechrate nach RIT von 91% ▲ eine komplette Remission nach RIT bei 69% der Patienten. Bei einem medianen Follow-up von 5,1 Jahren wurde ein progressionsfreies Überleben von 67% und ein Gesamtüberleben von 87% erreicht. Im Rahmen von Studien werden Konzepte mit Immunochemothe- rapie über nur drei Zyklen geprüft, auf die eine RIT-Konsolidation folgt (28). Chemotherapie Mono-Chemotherapien: Alkylanzien und Purinanaloga sind die am häufigsten verwendeten Monotherapien in der Behandlung der FL. Bei bis zu 3 von 4 behandelten Patienten kann mit diesen Monosubstanzen ein bedeutsames Ansprechen erzielt werden (29). Die Vorteile der Behandlung mit Alkylanzien (bspw. mit Chlorambucil oder Cyclophosphamid) sind die einfache Verabreichungsart, die gute Verträglichkeit mit relativ wenigen Nebenwirkungen und die Tatsache, dass sie gelegentlich beim Rezidiv wieder erfolgreich eingesetzt werden können. Bei den Purinanaloga ist das Fludarabin die am häufigsten verwendete Substanz. In der Erstlinie können Ansprechraten im Sinne von «clinical benefit» (CR/PR/SD) von 30 bis 70% (30) und beim Rezidiv von bis zu 64% (31) erzielt werden. Chemotherapeutische Kombinationsbehandlungen (ohne Antikörper): Verschiedene Kombinationsbehandlungen wie beispielsweise mit Cyclophosphamid, Vincristin und Prednison (CVP) wurden gegenüber einer Monotherapie verglichen (32, 33). Die Zugabe eines Anthrazyklins wie im CHOP-Schema führt zu höheren Responseraten und mässiger Verbesserung des progressionsfreien Überlebens, aber nicht (mit Ausnahme der FL Grad 3) zur Verbesserung des Gesamtüberlebens (34, 35). Weitere Kombinationsbehandlungen mit Mitoxantron (MCP- und CNOP-Schema [36, 37]) als auch Fludarabin-haltige Kombinationsbehandlungen (FC und FCM [38, 39]) wurden als Erstlinientherapie geprüft; allerdings konnte das langfristige Outcome gegenüber einer initialen Zytostatikamonotherapie nicht entscheidend verbessert werden. Hochdosistherapie mit autologer Stammzellunterstützung Bei Patienten mit FL in erster Remission kann die konsolidierende Hochdosistherapie mit autologer Stammzellunterstützung (ASZT) die Remissionsdauer verlängern (40–42). Diesem Vorteil steht aber eine höhere Inzidenz von Zweitneoplasien insbesondere nach TBI (Total-body irradiation) gegenüber (43). Insgesamt vermochte sich die ASZT nach einer initialen Induktionstherapie nicht durchzusetzen. Beim Rezidiv des FL wurde in einer kleineren Studie mit 89 randomisierten und evaluierbaren Patienten das progressionsfreie wie auch das Gesamtüberleben durch die ASZT signifikant verlängert. Zum Zeitpunkt der Studie waren Antikörperbe- 26 ONKOLOGIE 4/2008 Im Fokus: Maligne Lymphome und multiples Myelom handlungen aber noch nicht in der Klinik eingeführt. Der Stellenwert der ASZT beim Rezidiv bleibt deshalb unklar (44). Immunotherapien Interferon-alpha (INF-␣): INF-␣ wurde sowohl in Kombination mit Chemotherapie als auch als Einzelsubstanz in der Erhaltungstherapie geprüft. Die Studienresultate sind teilweise widersprüchlich. In einigen Studien konnte eine Verlängerung der Remissionsdauer und des Gesamtüberlebens erzielt werden, doch scheint diese Wirkung vor allem mit der Intensität der verwendeten Chemotherapie und mit derjenigen des INF-␣ gekoppelt zu sein. INF-␣ trägt weder zu einer höheren Ansprechrate noch zur Verbesserung des Gesamtüberlebens in der Erhaltungstherapie bei (45). Mit dem Aufkommen von Rituximab hat INF-␣ in der Klinik, vor allem wegen der beträchtlichen Nebenwirkungen, seine Bedeutung weitgehend verloren. Rituximab: Rituximab ist ein Anti-CD20-gerichteter monoklonaler Antikörper. Das Antigen CD20 findet sich auf den allermeisten (> 90%) Lymphozyten. Die Wirkung beruht sowohl auf der Aktivierung des Komplements als auch auf einer antikörpervermittelten zellulären Zytotoxizität. Rituximab wird in der Regel sehr gut toleriert. Beim Rezidiv des FL werden mit der Rituximab-Monotherapie Ansprechraten um 50% mit einer Remissionsdauer von rund 13 Monaten erzielt (46, 47). In der Erstlinienbehandlung steigt die Ansprechrate mit dem Standardregime (d.h. 4 Applikationen Rituximab 375 mg/m2 in wöchentlichem Abstand) auf 70 bis 75%, und es resultiert ein medianer Gewinn von 18 bis 24 Monaten bis zum Fortschreiten der Krankheit (48–51). Beim Rezidiv führt sowohl nach der Chemotherapie CHOP als auch nach CHOP plus Rituximab die Erhaltungstherapie mit Rituximab, in dreimonatigen Abständen über zwei Jahre, zu einer signifikanten Verbesserung des progressionsfreien Überlebens von 23 auf 52 Monate (für RCHOP vorbehandelte Patienten) und verbessert das Gesamtüberleben nach drei Jahren signifikant von 77% auf 85% (Zeitpunkt ab Randomisierung Rituximab versus keine Erhaltungstherapie) (52).
Die noch ausstehenden Daten der PRIMA-Studie werden die Frage definitiv klären, ob die Erhaltungstherapie mit Rituximab bereits nach der Erstlinientherapie von bedeutsamem Nutzen ist. Die in präklinischen Untersuchungen erzielten Resultate mit neuen Antikörpern geben Anlass zu Hoffnung, dass Rituximab durch noch wirksamere Substanzen ersetzt werden kann (53).
Chemoimmunotherapie Verschiedene Phase-III-Studien haben den Nutzen einer Kombinationsbehandlung von Rituximab mit einer klassischen Chemotherapie belegt. Praktisch immer kann die Ansprechrate, das krankheitsfreie Überleben und nach längerer Beobachtungszeit zunehmend auch ein verbessertes Gesamtüberleben dokumentiert werden. Zwei Metaanalysen, die eine mit 1943 Patienten und die andere mit 438 untersuchten Patienten, untermauern den Nutzen von Rituximab in Kombination mit Chemotherapie auf das Gesamtüberleben (54, 55). Ohne direkten Vergleich zwischen einer Polychemotherapie mit respektive ohne Anthrazyklin bleibt die Wahl der besten Chemotherapie in Kombination mit Rituximab® eine ungeklärte Frage. In Tabelle 3 sind wichtige Vergleichstudien mit Rituximab-Chemotherapie versus alleinige Chemotherapie aufgeführt.
Im Rahmen von Studien werden neue multimodale Behandlungskonzepte mit Konsolidierung durch Radio-Immunound Hochdosistherapie geplant. Offen ist, ob eine alleinige Antikörpertherapie mit Rituximab im langfristigen Verlauf einer Immunochemotherapie gefolgt von einer Rituximab-Erhaltungstherapie ebenbürtig ist. Die SAKK-35-08Studie (in Planung) wird diese Frage prüfen.
Allogene Transplantation Die allogene Stammzelltransplantation wird vor allem beim rezidivierenden und/oder refraktären FL diskutiert. Der Graft-versus-Lymphoma-Effekt kann zu einer langdauernden Remission führen. Da diese Behandlungsmodalität ein hohes Mortalitätsrisiko aufweist, ist die Indikation hierfür kritisch abzuwägen. ▲
Dr. med. Walter Mingrone Onkologische Abteilung Medizinische Klinik Kantonsspital Olten 4600 Olten E-Mail: wmingrone_ol@spital.ktso.ch
und
Dr. med. Alden Moccia Istituto Oncologico della Svizzera Italiana (IOSI) Ambulatorio di Oncologia Ospedale La Carità 6601 Locarno E-Mail: alden.moccia@eoc.ch
Merkpunkte
▲ In der Klinik ist der FLIPI-Score ein einfach zu bestimmender und verlässlicher prognostischer Parameter. Mit dem besseren Verständnis der Biologie wird in Zukunft mit grosser Wahrscheinlichkeit eine akkuratere Diagnose, Prognose und individualisiertere Therapie als bis anhin möglich sein.
▲ Der verzögerte Behandlungsbeginn (d.h. erst beim Auftreten von Symptomen oder raschem Progress) hat nicht notwendigerweise einen ungünstigen Einfluss auf die Gesamtlebenszeit. Beschwerdefreie Patienten mit einem indolenten und langsamen Verlauf können zunächst mit dem «watchful waiting» betreut werden.
▲ Immunochemotherapien mit Rituximab führen zu einer höheren Ansprechrate und verlängern das Gesamtüberleben. Bei therapiebedürftigen Patienten ist die Immunochemotherapie heute die Therapie der Wahl. Der Stellenwert der Anthrazyklin-haltigen Erstlinientherapie gegenüber einem Regime ohne Anthrazyklin bleibt offen.
▲ Zeitpunkt und optimale Dauer der Erhaltungstherapie mit Rituximab sind Gegenstand von Studien (bspw. SAKK 35/03). In der Schweiz ist die Kostenübernahme durch die Krankenkassen für Rituximab als Erhaltungstherapie (nach einer vorgängigen Polychemotherapie des Rezidivs) zurzeit auf zwei Jahre begrenzt.
▲ Radioimmunotherapien sind Erfolg versprechend und erweitern die therapeutischen Möglichkeiten. Der optimale Einsatz ist Gegenstand der klinischen Forschung.
ONKOLOGIE 4/2008
27
Im Fokus: Maligne Lymphome und multiples Myelom
Quellen:
(Die vollständige Titelliste kann über den Erstautor oder über die Redaktion angefordert werden.)
1. Morton LM et al.: Blood 2006; 107: 265–276.
2. Sandin S et al.: Cancer Epidemiol Biomarkers Prev 2006; 15(7): 1295–300.
3. D'Amore F et al.: Ann Oncol 2008; 19(4): iv131.
4. Zhang Y et al.: Americ. Epidemiol. 2008 167(11): 1321–1331.
5. Wang SS et al.: Blood 2007; 109: 3479–3488.
6. Yang E, Korsmeyer SJ: Blood 1996; 88(2): 386– 401.
7. Jaffe ES et al. (Eds). WHO Classification of Tumours. International Agency for Research on Cancer Press, Lyon 2001; 162–168.
8. Martinez A et al.: Arch Pathol Lab Med. 2007; 131: 1084–1088.
9. Wahlin B et al.: Clin Cancer Res 2007; 13(2): 388–397.
10. Carreras J et al.: Blood. 2006; 108: 2957–2964.
11. Ai WYZ et al.: Blood. 2006; 108; 824a.
12. Canioni D et al.: J Clin Oncol 2008; 26: 440– 446.
13. Horning SJ: Semin Oncol 1993; 20 (Suppl 5): 75–88.
14. Cheson B et al.: J Clin Oncol 2007; 25: 579–586.
15. Solal-Celigny P et al.: Blood 2004; 104: 1258– 1265.
16. Buske C et al.: Blood 2006; 108: 1504–8.
17. Wilder RB et al.: Int J Radiat Oncol Biol Phys 2001; 51: 1219–27.
18. Advani R et al.: J Clin Oncol 2004; 22: 1454–9. 19. Biermann PJ: Curr Opin Oncol 2007; 19: 433– 437. 20. Lopez-Guillermo A et al.: Blood 1999; 93: 3081–7. 21. Wilder RB et al.: Int J Radiat Oncol Biol Phys. 2001; 51: 1219–1227. 22. Haas R et al.: J Clin Oncol 2003; Vol 21: 2474– 2480. 23. Vose JM et al.: J Clin Oncol 2000; 18: 1316–23. 24. Witzig TE et al.: J Clin Oncol 2002; 20: 3262–9. 25. Karninski M et al.: NEJM 2005; 352: 441–449. 26. Press O et al.: J Clin Oncol 2006; 24: 4143–4149. 27. Hagenbeeck A et al.: Ann Oncol 2008; 19: iv181. 28. http://public.ukcrn.org.uk/search/StudyDetail. aspx?StudyID=4192. 29. Johnson, PW et al.: J Clin Oncol 1995; 13: 140–7. 30. Hagenbeek A et al.: Blood 1981; 58: 920–25. 31. Klasa RJ et al.: J Clin Oncol 2002; 20: 4649–54. 32. Lister A et al.: BMJ 1978; 1: 533–41. 33. Luce J et al.: Cancer 1971; 28: 306–17. 34. Dana BW et al.: J Clin Oncol 1993; 11: 644–51. 35. Ganti AK et al.: Ann Oncol 2006 17: 920–927. 36. Nickenig C et al.: Cancer 2006; 107: 1014–22. 37. Rohatgi N et al.: Am J Hematol 2002; 70: 181–5. 38. Hochster HS et al.: J Clin Oncol 2000; 18: 987–94. 39. Bosch F et al.: Blood 1997; 90: 530a. 40. Lenz G et al.: Blood 2004; 104: 2667–74. 41. Deconinck E et al.: Blood 2005; 105: 3817–23.
42. Sebban C et al.: Blood 2006; 108: 2540–4.
43. Montoto S et al.: Leukemia 2007; 21: 2324–2331.
44. Schouten HC et al.: J Clin Oncol 2003; 21: 3918–3927.
45. Rohatiner AZ et al.: J Clin Oncol 2005; 23: 2215– 23.
46. Davis TA et al.: J Clin Oncol 2000; 18: 3135–43.
47. Laughlin P et al.: J Clin Oncol 1998; 16: 2825– 2835.
48. Hainsworth JD et al.: Blood. 2000; 95: 3052– 3056.
49. Colombat P et al.: Blood. 2001; 97: 101–106.
50. Ghielmini M et al.: Blood. 2004; 103: 4416–4423.
51. Witzig TE et al.: J Clin Oncol. 2005; 23: 1103– 1108.
52. Van Oers M et al.: Blood 2006 108: 3295–3301.
53. Stanglmaier M et al.: Int J Cancer. 2008; 123(5): 1181–1189.
54. Schulz H et al.: J Natl Cancer Institute. 2007; 99: 706–714.
55. Sacchi S et al.: Cancer 2007; 109: 2077–2082.
56. Herold M et al.: Blood 2004; 104(11): 169a.
57. Salles G et al.: Blood 2004; 104(11): 49.
58. Marcus R et al.: Blood 2005; 105(4): 1417–23.
59. Hiddemann W et al.: Blood 2005; 106(12): 3725– 3732.
60. Van Oers MH et al.: Blood 2006; 108(10): 3295– 301.
61. Forstpointner R et al.: Blood 2004; 104(10): 3064–3071.
28 ONKOLOGIE 4/2008