

Transkript
Im Fokus: Maligne Lymphome und multiples Myelom
Extranodale Lymphome
Besonderheiten der Einteilung und Behandlung Neue biologische und klinische Erkenntnisse haben dazu geführt, dass extranodale Lymphome heute ganz anders behandelt werden als noch vor wenigen Jahren. Vor allem wurde gelernt, dass die Behandlungsstrategie sowohl die histologische Typisierung als auch die Charakteristika der Primärlokalisation berücksichtigen muss. Eine allgemeine Behandlungsstrategie für alle extranodalen Lymphome ist obsolet geworden.
PATRIZIA FRÖSCH, EMANUELE ZUCCA
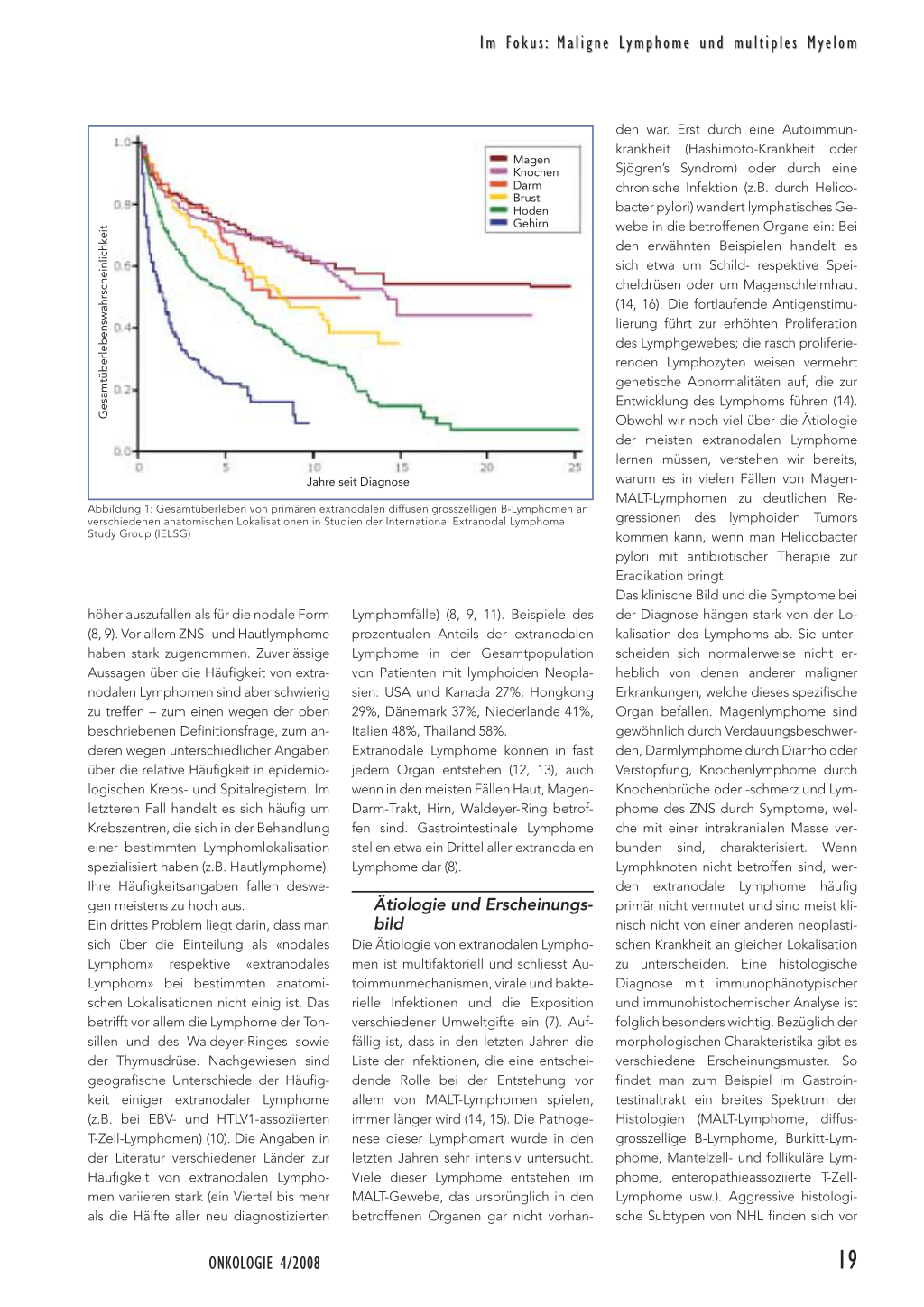
Die Bezeichnung «extranodale Lymphome» umfasst eine vielfältige Ansammlung verschiedener Morphologien, molekularer Abnormalitäten und klinischer Manifestationen. Die korrekte Diagnose und die regelrechte Behandlung der extranodalen Lymphome werden häufig durch diese Vielzahl der klinischen Bilder sowie durch die Seltenheit einiger dieser Lymphome deutlich erschwert. Somit kann kein einziges Krebszentrum allein eine genügende Erfahrung zum natürlichen Verlauf der extranodalen Lymphome an allen Lokalisationen sammeln. Wie aus Abbildung 1 ersichtlich ist, bildet die Lokalisation des extranodalen Lymphoms einen sehr wichtigen prognostischen Faktor. Das Istituto Oncologico della Svizzera Italiana (IOSI) hat vor mehr als zehn Jahren entschieden, die International Extranodal Lymphoma Study Group (IELSG) zu gründen, weil es nur innerhalb einer solchen internationalen, kooperativen Studiengruppe möglich ist, die nötige Anzahl von Fällen bei jeder Lymphomlokalisation zu sammeln, um Aussagen über den natürlichen Verlauf dieser speziellen Lymphomaarten machen zu können (1).
Diskussionspunkt Definition
Lymphome, die aus schleimhautassoziiertem lymphoidem Gewebe (= Mucose Associated Lymphoid Tissue, MALT) entstehen, stellen einen beträchtlichen Teil der extranodalen Lymphome dar. Diese MALT-Lymphome wurden erst durch P. Isaacson Mitte der Achtzigerjahre beschrieben (2) und blieben somit sehr lange in den gängigen Klassifikationen der NonHodkgin-Lymphome (NHL) unberücksichtigt. Erst mit dem Vorschlag der REAL-Klassifikation im Jahr 1994 (3) und danach in der WHO-Klassifikation (4) wurden sie berücksichtigt. Das heisst aber auch, dass ältere Studien und, um es einfach zu sagen, fast die gesamte Lymphomliteratur bis zur Jahrhundertwende
kaum brauchbar sind, um verlässliche Aussagen über Eigenschaften und Häufigkeit von extranodalen Lymphomen zu machen. Die genaue Definition des extranodalen Lymphoms bleibt auch noch heutzutage etwas unklar. Unter den verschiedenen Definitionen, die vorgeschlagen wurden, erachten wir diejenige von D’Amore für die brauchbarste (5). Diese sagt, dass man ein Lymphom als extranodal betrachtet, wenn die Hauptlokalisation der lymphoiden Neoplasie, die man behandeln muss, extranodal gelegen ist. Dabei muss die nodale Komponente (nodal = Lymphknoten oder Milz) weniger als 25% der gesamten Tumormasse ausmachen. Bei der Diagnosestellung befinden sich mehr als 80% aller Fälle von extranodalen Lymphomen in einem Stadium I oder II. Es gibt aber Autoren, die bestreiten, dass man den Begriff «extranodales Lymphom» für solche Fälle gebrauchen darf, in denen bei Diagnosestellung bereits ein Stadium III oder IV vorliegt. Wir stimmen dieser Position nicht zu und setzen voraus, dass man die zuvor erwähnte Definition von D’Amore richtig einsetzt. Es bleibt zu erwähnen, dass bei den meisten extranodalen Lymphomen bei der Stadieneinteilung die klassische Ann-Arbor-Terminologie angewandt wird, wie es heute bei fast allen Lymphomarten der Fall ist. In einigen Fällen ist eine spezielle Stadieneinteilung vorzuziehen, insbesondere bei Lymphomen des Gastrointestinaltrakts (GI-Lymphome), bei denen die sogenannte Lugano-Klassifikation nützlicher als das herkömmliche Ann-Arbor-System ist (6).
Diskussionspunkt Häufigkeit
Die Inzidenz von NHL hat sich in den westlichen Ländern in den letzten 40 Jahren etwas mehr als verdoppelt (7). Diese erstaunliche Zunahme scheint für extranodale lymphoide Neoplasien noch deutlich
18 ONKOLOGIE 4/2008
Im Fokus: Maligne Lymphome und multiples Myelom
Magen Knochen Darm Brust Hoden Gehirn
Gesamtüberlebenswahrscheinlichkeit
Jahre seit Diagnose
Abbildung 1: Gesamtüberleben von primären extranodalen diffusen grosszelligen B-Lymphomen an verschiedenen anatomischen Lokalisationen in Studien der International Extranodal Lymphoma Study Group (IELSG)
höher auszufallen als für die nodale Form (8, 9). Vor allem ZNS- und Hautlymphome haben stark zugenommen. Zuverlässige Aussagen über die Häufigkeit von extranodalen Lymphomen sind aber schwierig zu treffen – zum einen wegen der oben beschriebenen Definitionsfrage, zum anderen wegen unterschiedlicher Angaben über die relative Häufigkeit in epidemiologischen Krebs- und Spitalregistern. Im letzteren Fall handelt es sich häufig um Krebszentren, die sich in der Behandlung einer bestimmten Lymphomlokalisation spezialisiert haben (z.B. Hautlymphome). Ihre Häufigkeitsangaben fallen deswegen meistens zu hoch aus. Ein drittes Problem liegt darin, dass man sich über die Einteilung als «nodales Lymphom» respektive «extranodales Lymphom» bei bestimmten anatomischen Lokalisationen nicht einig ist. Das betrifft vor allem die Lymphome der Tonsillen und des Waldeyer-Ringes sowie der Thymusdrüse. Nachgewiesen sind geografische Unterschiede der Häufigkeit einiger extranodaler Lymphome (z.B. bei EBV- und HTLV1-assoziierten T-Zell-Lymphomen) (10). Die Angaben in der Literatur verschiedener Länder zur Häufigkeit von extranodalen Lymphomen variieren stark (ein Viertel bis mehr als die Hälfte aller neu diagnostizierten
Lymphomfälle) (8, 9, 11). Beispiele des prozentualen Anteils der extranodalen Lymphome in der Gesamtpopulation von Patienten mit lymphoiden Neoplasien: USA und Kanada 27%, Hongkong 29%, Dänemark 37%, Niederlande 41%, Italien 48%, Thailand 58%. Extranodale Lymphome können in fast jedem Organ entstehen (12, 13), auch wenn in den meisten Fällen Haut, MagenDarm-Trakt, Hirn, Waldeyer-Ring betroffen sind. Gastrointestinale Lymphome stellen etwa ein Drittel aller extranodalen Lymphome dar (8).
Ätiologie und Erscheinungsbild
Die Ätiologie von extranodalen Lymphomen ist multifaktoriell und schliesst Autoimmunmechanismen, virale und bakterielle Infektionen und die Exposition verschiedener Umweltgifte ein (7). Auffällig ist, dass in den letzten Jahren die Liste der Infektionen, die eine entscheidende Rolle bei der Entstehung vor allem von MALT-Lymphomen spielen, immer länger wird (14, 15). Die Pathogenese dieser Lymphomart wurde in den letzten Jahren sehr intensiv untersucht. Viele dieser Lymphome entstehen im MALT-Gewebe, das ursprünglich in den betroffenen Organen gar nicht vorhan-
den war. Erst durch eine Autoimmunkrankheit (Hashimoto-Krankheit oder Sjögren’s Syndrom) oder durch eine chronische Infektion (z.B. durch Helicobacter pylori) wandert lymphatisches Gewebe in die betroffenen Organe ein: Bei den erwähnten Beispielen handelt es sich etwa um Schild- respektive Speicheldrüsen oder um Magenschleimhaut (14, 16). Die fortlaufende Antigenstimulierung führt zur erhöhten Proliferation des Lymphgewebes; die rasch proliferierenden Lymphozyten weisen vermehrt genetische Abnormalitäten auf, die zur Entwicklung des Lymphoms führen (14). Obwohl wir noch viel über die Ätiologie der meisten extranodalen Lymphome lernen müssen, verstehen wir bereits, warum es in vielen Fällen von MagenMALT-Lymphomen zu deutlichen Regressionen des lymphoiden Tumors kommen kann, wenn man Helicobacter pylori mit antibiotischer Therapie zur Eradikation bringt. Das klinische Bild und die Symptome bei der Diagnose hängen stark von der Lokalisation des Lymphoms ab. Sie unterscheiden sich normalerweise nicht erheblich von denen anderer maligner Erkrankungen, welche dieses spezifische Organ befallen. Magenlymphome sind gewöhnlich durch Verdauungsbeschwerden, Darmlymphome durch Diarrhö oder Verstopfung, Knochenlymphome durch Knochenbrüche oder -schmerz und Lymphome des ZNS durch Symptome, welche mit einer intrakranialen Masse verbunden sind, charakterisiert. Wenn Lymphknoten nicht betroffen sind, werden extranodale Lymphome häufig primär nicht vermutet und sind meist klinisch nicht von einer anderen neoplastischen Krankheit an gleicher Lokalisation zu unterscheiden. Eine histologische Diagnose mit immunophänotypischer und immunohistochemischer Analyse ist folglich besonders wichtig. Bezüglich der morphologischen Charakteristika gibt es verschiedene Erscheinungsmuster. So findet man zum Beispiel im Gastrointestinaltrakt ein breites Spektrum der Histologien (MALT-Lymphome, diffusgrosszellige B-Lymphome, Burkitt-Lymphome, Mantelzell- und follikuläre Lymphome, enteropathieassoziierte T-ZellLymphome usw.). Aggressive histologische Subtypen von NHL finden sich vor
ONKOLOGIE 4/2008
19
Im Fokus: Maligne Lymphome und multiples Myelom
Gesamtüberlebenswahrscheinlichkeit
Stadium I –II (N= 131)
Stadium IV, multiple Schleimhautlokalisationen (N= 9)
Stadium IV, einschliesslich Knochenmark- und nodaler Erkrankung
Jahre seit Diagnose
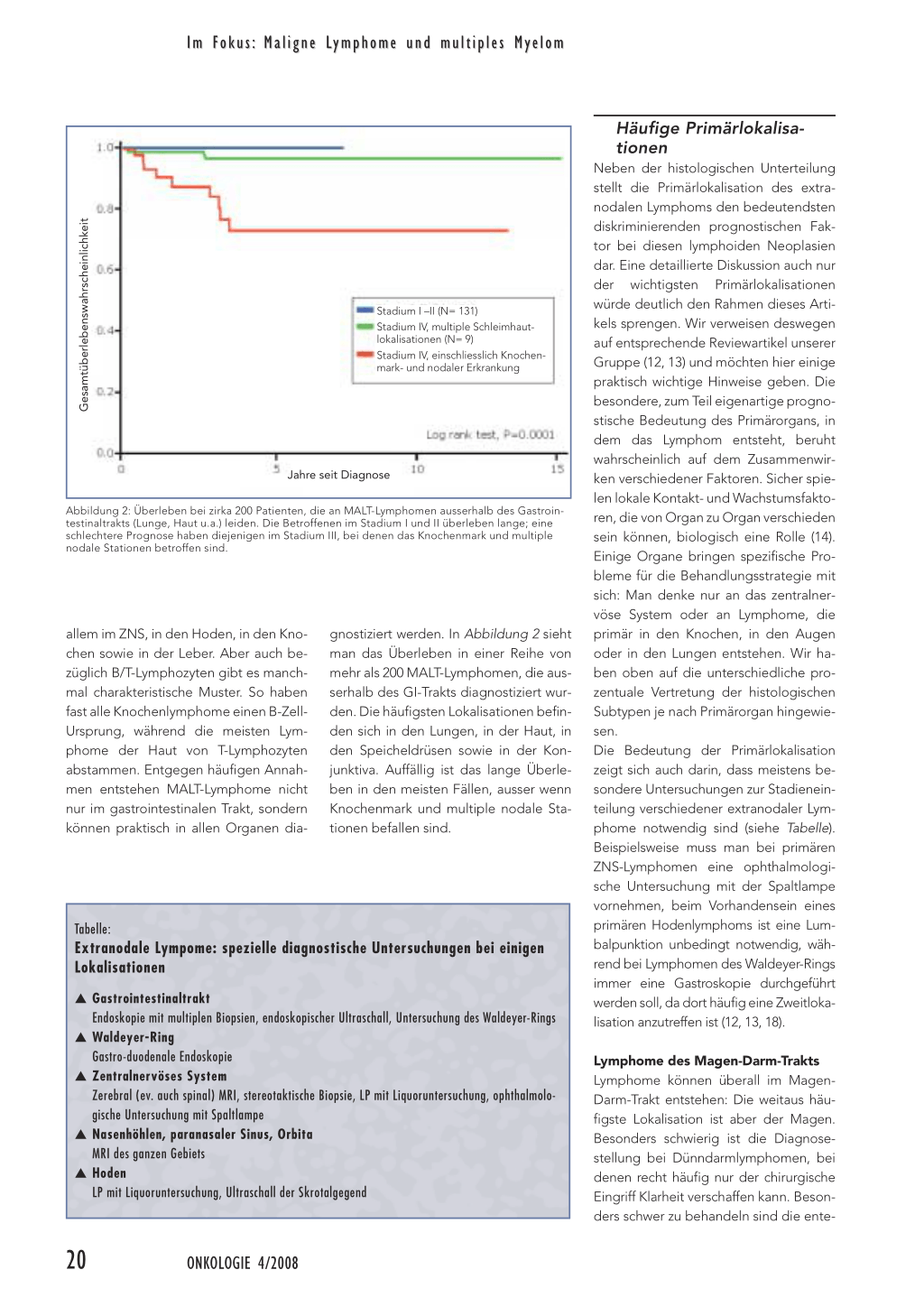
Abbildung 2: Überleben bei zirka 200 Patienten, die an MALT-Lymphomen ausserhalb des Gastrointestinaltrakts (Lunge, Haut u.a.) leiden. Die Betroffenen im Stadium I und II überleben lange; eine schlechtere Prognose haben diejenigen im Stadium III, bei denen das Knochenmark und multiple nodale Stationen betroffen sind.
allem im ZNS, in den Hoden, in den Knochen sowie in der Leber. Aber auch bezüglich B/T-Lymphozyten gibt es manchmal charakteristische Muster. So haben fast alle Knochenlymphome einen B-ZellUrsprung, während die meisten Lymphome der Haut von T-Lymphozyten abstammen. Entgegen häufigen Annahmen entstehen MALT-Lymphome nicht nur im gastrointestinalen Trakt, sondern können praktisch in allen Organen dia-
gnostiziert werden. In Abbildung 2 sieht man das Überleben in einer Reihe von mehr als 200 MALT-Lymphomen, die ausserhalb des GI-Trakts diagnostiziert wurden. Die häufigsten Lokalisationen befinden sich in den Lungen, in der Haut, in den Speicheldrüsen sowie in der Konjunktiva. Auffällig ist das lange Überleben in den meisten Fällen, ausser wenn Knochenmark und multiple nodale Stationen befallen sind.
Tabelle:
Extranodale Lympome: spezielle diagnostische Untersuchungen bei einigen Lokalisationen
▲ Gastrointestinaltrakt Endoskopie mit multiplen Biopsien, endoskopischer Ultraschall, Untersuchung des Waldeyer-Rings
▲ Waldeyer-Ring Gastro-duodenale Endoskopie
▲ Zentralnervöses System Zerebral (ev. auch spinal) MRI, stereotaktische Biopsie, LP mit Liquoruntersuchung, ophthalmologische Untersuchung mit Spaltlampe
▲ Nasenhöhlen, paranasaler Sinus, Orbita MRI des ganzen Gebiets
▲ Hoden LP mit Liquoruntersuchung, Ultraschall der Skrotalgegend
Häufige Primärlokalisationen
Neben der histologischen Unterteilung stellt die Primärlokalisation des extranodalen Lymphoms den bedeutendsten diskriminierenden prognostischen Faktor bei diesen lymphoiden Neoplasien dar. Eine detaillierte Diskussion auch nur der wichtigsten Primärlokalisationen würde deutlich den Rahmen dieses Artikels sprengen. Wir verweisen deswegen auf entsprechende Reviewartikel unserer Gruppe (12, 13) und möchten hier einige praktisch wichtige Hinweise geben. Die besondere, zum Teil eigenartige prognostische Bedeutung des Primärorgans, in dem das Lymphom entsteht, beruht wahrscheinlich auf dem Zusammenwirken verschiedener Faktoren. Sicher spielen lokale Kontakt- und Wachstumsfaktoren, die von Organ zu Organ verschieden sein können, biologisch eine Rolle (14). Einige Organe bringen spezifische Probleme für die Behandlungsstrategie mit sich: Man denke nur an das zentralnervöse System oder an Lymphome, die primär in den Knochen, in den Augen oder in den Lungen entstehen. Wir haben oben auf die unterschiedliche prozentuale Vertretung der histologischen Subtypen je nach Primärorgan hingewiesen. Die Bedeutung der Primärlokalisation zeigt sich auch darin, dass meistens besondere Untersuchungen zur Stadieneinteilung verschiedener extranodaler Lymphome notwendig sind (siehe Tabelle). Beispielsweise muss man bei primären ZNS-Lymphomen eine ophthalmologische Untersuchung mit der Spaltlampe vornehmen, beim Vorhandensein eines primären Hodenlymphoms ist eine Lumbalpunktion unbedingt notwendig, während bei Lymphomen des Waldeyer-Rings immer eine Gastroskopie durchgeführt werden soll, da dort häufig eine Zweitlokalisation anzutreffen ist (12, 13, 18).
Lymphome des Magen-Darm-Trakts Lymphome können überall im MagenDarm-Trakt entstehen: Die weitaus häufigste Lokalisation ist aber der Magen. Besonders schwierig ist die Diagnosestellung bei Dünndarmlymphomen, bei denen recht häufig nur der chirurgische Eingriff Klarheit verschaffen kann. Besonders schwer zu behandeln sind die ente-
20 ONKOLOGIE 4/2008
Im Fokus: Maligne Lymphome und multiples Myelom
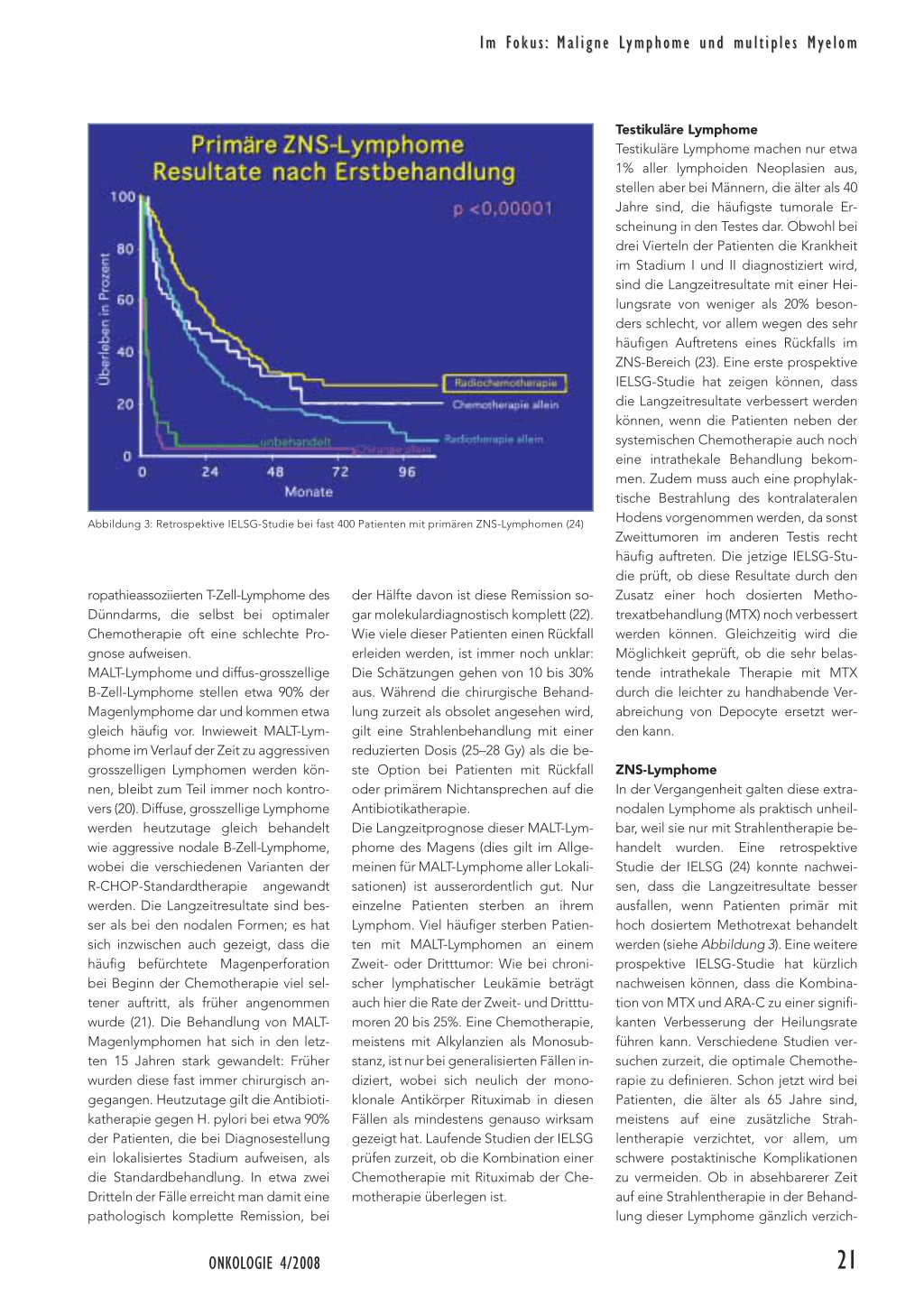
Abbildung 3: Retrospektive IELSG-Studie bei fast 400 Patienten mit primären ZNS-Lymphomen (24)
ropathieassoziierten T-Zell-Lymphome des Dünndarms, die selbst bei optimaler Chemotherapie oft eine schlechte Prognose aufweisen. MALT-Lymphome und diffus-grosszellige B-Zell-Lymphome stellen etwa 90% der Magenlymphome dar und kommen etwa gleich häufig vor. Inwieweit MALT-Lymphome im Verlauf der Zeit zu aggressiven grosszelligen Lymphomen werden können, bleibt zum Teil immer noch kontrovers (20). Diffuse, grosszellige Lymphome werden heutzutage gleich behandelt wie aggressive nodale B-Zell-Lymphome, wobei die verschiedenen Varianten der R-CHOP-Standardtherapie angewandt werden. Die Langzeitresultate sind besser als bei den nodalen Formen; es hat sich inzwischen auch gezeigt, dass die häufig befürchtete Magenperforation bei Beginn der Chemotherapie viel seltener auftritt, als früher angenommen wurde (21). Die Behandlung von MALTMagenlymphomen hat sich in den letzten 15 Jahren stark gewandelt: Früher wurden diese fast immer chirurgisch angegangen. Heutzutage gilt die Antibiotikatherapie gegen H. pylori bei etwa 90% der Patienten, die bei Diagnosestellung ein lokalisiertes Stadium aufweisen, als die Standardbehandlung. In etwa zwei Dritteln der Fälle erreicht man damit eine pathologisch komplette Remission, bei
der Hälfte davon ist diese Remission sogar molekulardiagnostisch komplett (22). Wie viele dieser Patienten einen Rückfall erleiden werden, ist immer noch unklar: Die Schätzungen gehen von 10 bis 30% aus. Während die chirurgische Behandlung zurzeit als obsolet angesehen wird, gilt eine Strahlenbehandlung mit einer reduzierten Dosis (25–28 Gy) als die beste Option bei Patienten mit Rückfall oder primärem Nichtansprechen auf die Antibiotikatherapie. Die Langzeitprognose dieser MALT-Lymphome des Magens (dies gilt im Allgemeinen für MALT-Lymphome aller Lokalisationen) ist ausserordentlich gut. Nur einzelne Patienten sterben an ihrem Lymphom. Viel häufiger sterben Patienten mit MALT-Lymphomen an einem Zweit- oder Dritttumor: Wie bei chronischer lymphatischer Leukämie beträgt auch hier die Rate der Zweit- und Dritttumoren 20 bis 25%. Eine Chemotherapie, meistens mit Alkylanzien als Monosubstanz, ist nur bei generalisierten Fällen indiziert, wobei sich neulich der monoklonale Antikörper Rituximab in diesen Fällen als mindestens genauso wirksam gezeigt hat. Laufende Studien der IELSG prüfen zurzeit, ob die Kombination einer Chemotherapie mit Rituximab der Chemotherapie überlegen ist.
Testikuläre Lymphome Testikuläre Lymphome machen nur etwa 1% aller lymphoiden Neoplasien aus, stellen aber bei Männern, die älter als 40 Jahre sind, die häufigste tumorale Erscheinung in den Testes dar. Obwohl bei drei Vierteln der Patienten die Krankheit im Stadium I und II diagnostiziert wird, sind die Langzeitresultate mit einer Heilungsrate von weniger als 20% besonders schlecht, vor allem wegen des sehr häufigen Auftretens eines Rückfalls im ZNS-Bereich (23). Eine erste prospektive IELSG-Studie hat zeigen können, dass die Langzeitresultate verbessert werden können, wenn die Patienten neben der systemischen Chemotherapie auch noch eine intrathekale Behandlung bekommen. Zudem muss auch eine prophylaktische Bestrahlung des kontralateralen Hodens vorgenommen werden, da sonst Zweittumoren im anderen Testis recht häufig auftreten. Die jetzige IELSG-Studie prüft, ob diese Resultate durch den Zusatz einer hoch dosierten Methotrexatbehandlung (MTX) noch verbessert werden können. Gleichzeitig wird die Möglichkeit geprüft, ob die sehr belastende intrathekale Therapie mit MTX durch die leichter zu handhabende Verabreichung von Depocyte ersetzt werden kann.
ZNS-Lymphome In der Vergangenheit galten diese extranodalen Lymphome als praktisch unheilbar, weil sie nur mit Strahlentherapie behandelt wurden. Eine retrospektive Studie der IELSG (24) konnte nachweisen, dass die Langzeitresultate besser ausfallen, wenn Patienten primär mit hoch dosiertem Methotrexat behandelt werden (siehe Abbildung 3). Eine weitere prospektive IELSG-Studie hat kürzlich nachweisen können, dass die Kombination von MTX und ARA-C zu einer signifikanten Verbesserung der Heilungsrate führen kann. Verschiedene Studien versuchen zurzeit, die optimale Chemotherapie zu definieren. Schon jetzt wird bei Patienten, die älter als 65 Jahre sind, meistens auf eine zusätzliche Strahlentherapie verzichtet, vor allem, um schwere postaktinische Komplikationen zu vermeiden. Ob in absehbarerer Zeit auf eine Strahlentherapie in der Behandlung dieser Lymphome gänzlich verzich-
ONKOLOGIE 4/2008
21
Im Fokus: Maligne Lymphome und multiples Myelom
tet werden kann, bleibt noch kontrovers. Randomisierte Studien, die diese Frage überprüfen, werden derzeit international geplant.
Weitere Lokalisationen Zwei retrospektive IELSG-Studien haben kürzlich nachgewiesen, dass die Langzeitresultate bei Knochenlymphomen und bei lymphoiden Neoplasien, die in der Brust primär entstehen, viel besser sind, als man früher annahm (25, 26). Auch hier stellt sich jetzt die Frage, ob die Resultate mit einer modernen systemischen Behandlung dieser Lymphome so gut werden, dass in absehbarer Zeit auf eine zusätzliche Strahlentherapie verzichtet werden kann. Eine andere wichtige retrospektive IELSG-Studie hat gezeigt, dass bei fast 450 Fällen von primär mediastalen Lymphomen (PMBL) intensivere Chemotherapien der Standardkombination CHOP überlegen sind, was bei nodalen Lymphomen bekanntlich nicht der Fall ist (27). Laufende Studien versuchen zurzeit, die Rolle von Rituximab und einer zusätzlichen Strahlentherapie bei dieser besonderen Lymphomart zu klären. Interessanterweise haben wir ja in der Zwischenzeit gelernt, dass das PMBL in der Grauzone zwischen NHL und Hodgkin-Lymphomen zu positionieren ist (28). Dies erklärt wohl auch, warum die Langzeitresultate beim PMBL deutlich besser als bei «gewöhnlichen» diffusen nodalen grosszelligen Lymphomen ausfallen.
Zusammenfassung
Die jüngsten Erkenntnisse über extranodale Lymphome haben weitreichende
Folgen für die Therapieoptionen. Immer zu beachten ist, dass nicht einmal die grössten Krebszentren über eine genügend grosse Anzahl an Fällen verfügen, um bei jeder Primärlokalisation selbstständig Therapierichtlinien aufstellen zu können. Die vor fast 15 Jahren gegründete International Extranodal Lymphoma Study Group (IELSG, www.ielsg.org) ist zur weltführenden Arbeitsgruppe auf dem Gebiet dieser Lymphomart geworden. Der Arzt, der bei einem Patienten ein extranodales Lymphom vermutet, ist deswegen gut beraten, sich bei einem erfahrenen Kollegen aus diesem Team über die neusten diagnostischen und therapeutischen Optionen zu informieren. ▲
Dr. med. Patrizia Frösch (Korrespondenzadresse) Istituto Oncologico della Svizzera Italiana (IOSI) Ospedale Regionale Bellinzona e Valli 6500 Bellinzona E-Mail: froesch@ticino.com
und
PD Dr. med. Emanuele Zucca Istituto Oncologico della Svizzera Italiana (IOSI) 6500 Bellinzona
Quellen: (Die vollständige Titelliste kann bei der Erstautorin oder über die Redaktion angefordert werden.) 1. www.ielsg.org 2. Isaacson PG: Annals of Oncology 1995; 6: 319–320. 3. Harris NL et al.: Blood 1994; 84: 1361–1392. 4. Jaffe ES et al.: IARC Press, 2001: 1–351.
5. d’Amore F et al.: Eur J Cancer 1991; 27: 1201–8.
6. Rohatiner A et al.: Annals of Oncology 1994; 5: 397–407.
7. Parkin DM et al.: CA Cancer J Clin 2005; 55: 74–108.
8. Groves FD et al.: J Natl Cancer Inst 2000; 92: 1240–51.
9. Chiu BC et al.: Clin Lymphoma 2003; 4: 161–8.
10. Morton LM et al.: Blood 2006; 107: 265–76.
11. Economopoulos T et al.: Leuk Lymphoma 1996; 21: 131–6.
12. Zucca E et al.: Annals of Oncology 1997; 8: 727–737.
13. Zucca E et al.: Ann Oncol 1999; 10: 1023–33.
14. Bertoni F, Zucca E: J Clin Oncol 2005; 23: 6415–20.
15. Ferreri AJ et al.: Ann Oncol 2007.
16. Suarez F et al.: Blood 2006; 107: 3034–44.
17. Zucca E et al.: Blood 2003; 101: 2489–95.
18. Thieblemont C, Coiffier B. In: Zucca E, Bertoni F (eds.). MALT Lymphomas 2004: 60–80.
19. Sutcliffe SB, Gospodarowicz MK. In: Keating A et al. (eds.) Hematological Oncology. 1992: 189–222.
20. Zucca E et al.: Blood 2000; 96: 410–419.
21. Cortelazzo S et al.: Ann Oncol 1999; 10: 1433–40.
22. Pinotti G et al.: Leukemia and Lymphoma 1997; 26: 527–537.
23. Zucca E et al.: J Clin Oncol 2003; 21: 20–7.
24. Ferreri AJ et al.: J Clin Oncol 2003; 21: 266–72.
25. Christie D et al.: Haematologica 2007; 92 (Suppl 1): Abstract # 0717.
26. Ryan G et al.: Ann Oncol 2008; 19: 223–241.
27. Zinzani PL et al.: Haematologica 2002; 87: 1258–1264.
28. Rosenwald A et al.: J Exp Med 2003; 198: 851–862.
22 ONKOLOGIE 4/2008