





Transkript
Im Fokus: Maligne Lymphome und multiples Myelom
Chronische lymphatische Leukämie
Epidemiologie, Diagnostik, prognostische Marker,Therapie Nach langer Zeit ohne wesentliche diagnostische und therapeutische Fortschritte stehen heute bei der chronischen lymphatischen Leukämie (CLL) zahlreiche neue prognostische Marker und hochwirksame Kombinationstherapien zur Verfügung. Welche Untersuchungen sollen heute bei Patienten mit CLL im klinischen Alltag durchgeführt werden? Welche Patienten sind für die neueren, intensiveren Therapien geeignet?
MICHAEL GREGOR
Die CLL ist die häufigste Leukämieform in Europa und Nordamerika. Die Inzidenz beträgt 3 bis 4 pro 100 000 Einwohner pro Jahr. Sie steigt mit zunehmendem Alter kontinuierlich an und liegt bei 75Jährigen bei etwa 20/100 000/Jahr. Das mediane Alter bei Diagnose liegt bei 65 bis 70 Jahren. Knapp ein Drittel der Patienten ist bei Diagnose unter 60 Jahre alt, nur etwa 10% ist jünger als 50 Jahre. Männer erkranken beinahe doppelt so häufig wie Frauen. Die Ätiologie der CLL ist unbekannt. Einige Autoren berichten über eine Assoziation mit der Exposition gegenüber Herbiziden (Beobachtung bei Landarbeitern). Bei erstgradigen Verwandten von CLL-Patienten ist das Erkrankungsrisiko deutlich erhöht. Die Pathogenese der CLL ist nur in Grundzügen bekannt. Neben genetischen Faktoren spielen auch Umweltfaktoren und das Immunsystem eine Rolle. Die Verwendung einer beschränkten Anzahl von B-Zell-Rezeptoren deutet auf eine chronische Stimulation des Immunsystems mit nachfolgender Fehlregulierung desselben hin. Diese Fehlregulierung betrifft sowohl die B- wie auch die T-Lymphozyten und die Stromazellen. Erworbene, unterschiedliche, genetische Veränderungen der CLL-Zellen führen zu deren Akkumulation und Proliferation (1). Die CLL ist ein noch nicht vollständig verstandenes Modell für eine multifaktoriell bedingte Tumorentstehung.
Definition und Diagnose
Gemäss WHO-Klassifikation ist die CLL definiert als leukämische Form des lymphozytischen Lymphoms vom B-Zell-Typ. Morphologisch ähnliche T-Zell-Erkrankungen werden als T-Prolymphozyten-Leukämie (T-PLL) bezeichnet. Zur Diagnosestellung ist eine
Lymphozytose von > 5 G/l (> 5 x 109/l) während mindestens drei Monaten notwendig (Abbildung 1). Zur Unterscheidung der CLL von anderen leukämischen lymphoproliferativen Erkrankungen ist eine Immunphänotypisierung des Blutes notwendig. Die CLL-Lymphozyten exprimieren neben dem B-ZellOberflächenantigen CD19 typischerweise CD5 und CD23 sowie Immunglobulin-Leichtketten-Typ Kappa oder Lambda. Weitere B-Zell-Oberflächenantigene wie CD20, CD22, CD79b, IgM und FMC7 werden in der Regel nur schwach exprimiert. Das Blutbild und eine Immunphänotypisierung genügen in den meisten Fällen zur Diagnose einer CLL. Eine Knochenmarkuntersuchung oder eine Lymphknotenbiopsie sind zur Diagnosestellung nicht notwendig (Tabelle 1).
Klinik
Heute erfolgt die Diagnosestellung einer CLL bei zwei Dritteln bis drei Vierteln aller Patienten in einem asymptomatischen Frühstadium aufgrund einer Blutbilduntersuchung, welche aus einer anderen Indikation veranlasst wurde (z.B. Check-up, präoperativ, Infekt). Bei symptomatischen Patienten stehen unspezifische Anämiesymptome und eine Infektneigung im Vordergrund. Konstitutionelle Symptome wie Fieber, Nachtschweiss und Gewichtsverlust (BSymptome) sind als Erstsymptome selten. Ihr Auftreten sollte an die Möglichkeit einer Transformation in ein aggressives Lymphom denken lassen (RichterSyndrom). In fortgeschrittenen Stadien findet man bei der klinischen Untersuchung vergrösserte Lymphknoten und teilweise eine Splenomegalie oder Hepatomegalie.
6 ONKOLOGIE 4/2008
Im Fokus: Maligne Lymphome und multiples Myelom
Abbildung 1: Blutausstrich bei CLL Im Blutausstrich sieht man bei der CLL typischerweise kleine und reif wirkende Lymphozyten mit einem schmalen Zytoplasmasaum und einem dichten Zellkern ohne klar erkennbaren Nukleolus. Kernschatten oder Gumprecht’sche Schollen sind ein für ein CLL typisches Artefakt im manuell angefertigten Blutausstrich. Daneben gibt es Formen mit atypischer Morphologie, gebuchteten Zellen oder grossen Zellen mit deutlichem Nukleolus und weiterem Zytoplasma («Prolymphozyten»). Bei einem Anteil der Prolymphozyten > 55% wird die Erkrankung nicht mehr als CLL, sondern als Prolymphozyten-Leukämie (B-PLL) bezeichnet.
Staging und prognostische Marker
Die heute verwendeten Staging-Systeme, sowohl das in Nordamerika verbreitete nach Rai wie auch das in Europa üblichere nach Binet, beruhen auf einer einfachen klinischen Untersuchung und auf einem Blutbild (2, 3). Radiologische Untersuchungen werden dabei nicht berücksichtigt. Beide Klassifikationen können Patienten im Frühstadium mit guter Prognose von Patienten in fortgeschrittener Erkrankung mit schlechter Prognose unterscheiden (Tabelle 2). Sie sind allerdings statisch, das heisst, sie berücksichtigen lediglich die Tumorausdehnung und das Knochenmarkversagen und keine weiteren Faktoren, welche den Verlauf und das Therapieansprechen beeinflussen. In den vergangenen Jahren wurden zahlreiche prognostische Faktoren beschrieben, welche nur zum Teil in nachfolgenden Untersuchungen validiert werden konnten. Die meisten prognostischen Marker können in Frühstadien das Risiko einer Progression der CLL gut abschätzen. Sobald es zu einem Therapiebedarf kommt, ist deren Bedeutung jedoch deutlich geringer. Eine kurze Lymphozytenverdoppelungszeit (unter 12 Monaten) ist mit einer schlechteren Prognose assoziiert (4). Sie kann bei allen Patienten problemlos bei
den Kontrolluntersuchungen bestimmt werden. Eine Erhöhung der einfach messbaren Serumparameter Beta-2-Mikroglobulin und Thymidinkinase korreliert mit einem fortgeschrittenen Stadium und mit kürzerem progressionsfreiem Überleben. Beide Parameter behalten in Multivarianzanalysen eine unabhängige prognostische Information für progressionsfreies Überleben, aber nicht für das Gesamtüberleben (5). Die mittels Immunphänotypisierung auf den CLL-Zellen bestimmte Expression von CD38 (Cut-off-Werte je nach Studie 7%, 20% oder 30%) und ZAP70 (Cut-off 20%) ist mit einer schlechten Prognose assoziiert. Beide Parameter korrelieren teilweise mit unmutiertem Immunglobulingen-Mutationsstatus (IgVH) (6, 7). Bei rund 20% der Patienten findet man jedoch eine Diskrepanz zum Mutationsstatus. Für beide Parameter existieren keine validierten Standardmethoden für deren Bestimmung und für die Cut-offSchwelle. Der IgVH-Mutationsstatus der CLL-Lymphozyten hat eine relevante prognostische Bedeutung. CLL-Patienten mit unmutiertem IgVH-Rearrangement haben ein kürzeres progressionsfreies Überle-
ben und Gesamtüberleben als Patienten mit mutiertem IgVH-Rearrangement (8). Die Verwendung des VH3-21-Gens ist unabhängig vom Mutationsstatus mit einer schlechten Prognose assoziiert (9). Eine wesentliche prognostische Information liefert die zytogenetische Untersuchung der CLL-Lymphozyten mittels Fluoreszenz-in-situ-Hybridisierung (FISH). Mit dieser Technologie kann man bei rund 80% der CLL-Patienten chromosomale Aberrationen feststellen: Patienten mit Deletion des Kurzarms des Chromosoms 17 (del17p), weitgehend entsprechend einer Deletion des Tumorsuppressorgens p53, haben eine sehr schlechte Prognose (medianes Gesamtüberleben 30 Monate). Patienten mit Deletion des Langarms des Chromosoms 11 (del11q22) haben eine ungünstige Prognose (medianes Gesamtüberleben 84 Monate). Patienten mit Trisomie 12 oder normaler Zytogenetik haben eine intermediäre Prognose (medianes Gesamtüberleben jeweils 120 Monate). Patienten mit Deletion des Langarms des Chromosoms 13 als einzige Aberration (del13q14) haben eine gute Prognose (medianes Gesamtüberleben 132 Monate) (10). Der Nachweis einer del(17p) oder einer del (11q22) hat auch eine prädiktive Be-
Tabelle 1:
Diagnostische Untersuchung bei CLL im klinischen Alltag (nach 11)
Obligatorische Untersuchungen zur Diagnosesicherung ▲ Blutbild mit mikroskopischer Differenzierung ▲ Immunphänotypisierung der Lymphozyten
Obligatorische Untersuchungen vor Therapiebeginn ▲ Anamnese, klinische Untersuchung, Performance Status ▲ Blutbild mit mikroskopischer Differenzierung ▲ Laborchemische Untersuchungen, Immunglobuline ▲ direkter Antihumanglobulintest (Coombs-Test) ▲ Infektserologien (HIV, HBV, HCV, evtl. CMV) ▲ Röntgenuntersuchung des Thorax
Fakultative Untersuchungen vor Therapiebeginn ▲ Zytogenetik (FISH) aus peripherem Blut1 ▲ Ultraschall2- oder CT-Untersuchung3 des Abdomens
Nicht routinemässig indizierte Untersuchungen ▲ IgVH-Mutationsstatus, CD38, ZAP70 ▲ MRD-Bestimmung
Als klinischer Alltag gilt jede Behandlung ausserhalb von Studien. 1 Bei allen Patienten mit therapeutischer Konsequenz wünschenswert! 2 Sofern zur Beurteilung einer Hepatosplenomegalie notwendig. 3 Falls eine komplette Remission mit einer intensiven Therapie angestrebt wird.
ONKOLOGIE 4/2008
7
Im Fokus: Maligne Lymphome und multiples Myelom
Tabelle 2:
Staging nach Binet
Stadium Binet A Binet B Binet C
Definition < 3 Lymphknotenregionen vergrössert ≥ 3 Lymphknotenregionen vergrössert Anämie (Hämoglobin < 100 g/l) oder Thrombozytopenie (< 100 g/l) medianes Überleben (Jahre) > 10 5 3
Als Lymphknotenregionen zählen zervikale, axillare, inguinale Lymphknotenvergrösserungen, egal ob unilateral oder bilateral befallen, sowie Leber und Milz. Lymphknoten gelten ab einem palpatorischen Durchmesser über 1,5 cm als vergrössert. Zur Beurteilung einer Leber- und Milzvergrösserung wird lediglich der klinische Status verwendet. Radiologische Untersuchungen werden für das Staging nach Binet nicht berücksichtigt. Autoimmune Zytopenien und andere Ursachen für eine Anämie oder Thrombozytopenie sollten ausgeschlossen werden.
Tabelle 3:
Therapieindikationen nach IWCLL 2007 (11) :
1. Knochenmarkversagen mit Entwicklung oder Verschlechterung einer Anämie oder Thrombozytopenie 2. ausgeprägte (> 6 cm unter linkem Rippenbogen), zunehmende oder symptomatische Splenomegalie 3. ausgeprägte (> 10 cm Maximaldurchmesser), zunehmende oder symptomatische Lymphadenopathie 4. zunehmende Lymphozytose mit einem Anstieg von >50% innert 2 Monaten oder Lymphozytenver-
dopplungszeit < 6 Monaten; dabei Zurückhaltung bei Lympohozytenzahlen < 30 G/l (30 x 109/l). Sowohl bei zunehmender Lymphadenopathie wie auch bei der Lymphozytose ist der Ausschluss anderer Ursachen, insbesondere von Infektionen, notwendig. 5. autoimmunhämolytische Anämie und / oder autoimmune Thrombozytopenie ohne Ansprechen auf Standardtherapien 6. krankheitsbedingte Symtome (eines von a bis d): a) ungewollter Gewichtsverlust ≥ 10% innert 6 Monaten b) relevante Einschränkung der Leistungsfähigkeit (ECOG PS ≤ 2, Arbeitsunfähigkeit) c) Fieber > 38,0° C während ≥ 2 Wochen ohne Hinweise auf Infektion d) Nachtschweiss während ≥ 1 Monat ohne Hinweise auf Infektion
deutung. Patienten mit del(17p) sprechen auf eine Therapie mit Purinanaloga und Alkylantien nur schlecht an. Sie zeigen aber ein gutes Ansprechen auf eine Antikörpertherapie mit Alemtuzumab (11). Patienten mit del(11q22) sprechen auf Monotherapien mit Purinanaloga und Alkylantien nur schlecht an. Das Ansprechen auf eine Kombinationstherapie mit Fludarabin und Cyclophosphamid scheint deutlich besser, bei allerdings kurzer Ansprechdauer. Derzeit existiert kein allgemein gültiges prognostisches Scoringsystem für die CLL wie IPI und FLIPI für Lymphome. Einzig der zytogenetische Nachweis einer del(17p) oder einer del(11q22) mittels FISH hat Konsequenzen für das therapeutische Vorgehen. Deshalb wird für den klinischen Alltag beim Einsatz der oben kurz diskutierten prognostischen Marker Zurückhaltung empfohlen (Tabelle 1).
Therapieprinzipien der CLL
Therapieindikationen Asymptomatische Patienten in Frühstadien (Binet A) werden beobachtet und erst bei Krankheitsprogression oder Auftreten von Symptomen behandelt. Mehrere Studien haben gezeigt, dass eine Therapie mit Alkylanzien in Frühstadien das Überleben nicht verlängert. Studien mit wirksameren, intensiveren Therapien bei Patienten mit hohem Progressionsrisiko werden derzeit durchgeführt (CLL7Studie der DCLLSG). Patienten im intermediären Stadium (Binet B) können (ohne Therapie) beobachtet werden, bis die Krankheit fortschreitet oder symptomatisch wird. Patienten mit aktiver symptomatischer Erkrankung (unabhängig vom Stadium!) und die meisten Patienten im Stadium Binet C profitieren von einer Behandlung (Tabelle 3). Bei Patienten mit CLL tritt in rund 20% im
Verlauf eine autoimmunhämolytische Anämie (AIHA) und in etwa 3% eine autoimmune Thrombozytopenie (ITP) auf. Sowohl eine AIHA wie eine ITP sollten entsprechend den jeweiligen Therapierichtlinien primär mit Steroiden und nicht mit zytostatischer Therapie behandelt werden, ausser wenn gleichzeitig ein Therapiebedarf bezüglich der CLL vorliegt. Bei Steroidresistenz oder Rezidiv ist individuell abzuwägen, ob eine Zweitlinientherapie der AIHA oder ITP mit Immunglobulinen, Immunsuppressiva oder Splenektomie oder ob eine CLL-Therapie angezeigt ist (12). Bei Patienten mit aktiver AIHA ist eine Monotherapie mit Purinanaloga kontraindiziert. Eine hohe Leukozytenzahl und eine Hypogammaglobulinämie alleine gelten nicht als Indikation für eine zytostatische Therapie.
Untersuchungen vor Therapie Vor Therapieeinleitung müssen neben den erwähnen Untersuchungen zur Diagnosesicherung weitere zusätzliche Untersuchungen durchgeführt werden (Tabelle 1). Eine Knochenmarkuntersuchung dient der Abklärung einer Zytopenie und sollte vor jeder Therapie angestrebt werden. Eine Lymphknotenbiopsie ist erforderlich, falls eine Transformation in ein aggressives Lymphom vermutet wird (Richter-Syndrom). Weitere Untersuchungen sind zur Suche nach möglichen Komorbiditäten notwendig, welche die Therapiewahl einschränken könnten (z.B. Niereninsuffizienz). Wichtig sind auch die Virus- Serologien (entsprechend HIV, Hepatitis B, Hepatitis C und, bei Antikörpertherapie, CMV), da es besonders unter immunsuppressiv wirksamen Therapien zu einer Reaktivierung der Virusinfekte kommen kann.
Beurteilung des Therapieansprechens Wie bei jeder zytostatischen Therapie sollte auch bei CLL eine Beurteilung des Therapieansprechens erfolgen. Sie beruht in erster Linie auf einer klinischen und auf einer Blutuntersuchung. Eine Knochenmarkpunktion und bildgebende Verfahren sind ausserhalb von Studien nur sinnvoll, falls mit intensiveren Therapien eine komplette Remission angestrebt wird oder falls eine spezifische Fragestellung hierzu besteht (z.B. anhal-
8 ONKOLOGIE 4/2008
Im Fokus: Maligne Lymphome und multiples Myelom
tende Zytopenie nach Therapieabschluss) (Tabelle 4).
Minimal residuelle Erkrankung Mittels Immunphänotypisierung oder quantitativer PCR kann bei vielen Patienten mit kompletter Remission eine minimale residuelle Erkrankung nachgewiesen werden (MRD). Die Sensitivität der quantitativen MRD-Bestimmung liegt mit beiden Methoden bei 0,01% (eine CLL-Zelle auf 10 000 Leukozyten). MRDUntersuchungen können sowohl im Blut wie auch im Knochenmark durchgeführt werden, mit Ausnahme der ersten Monate nach Antikörpertherapien. Patienten ohne MRD haben ein besseres progressionsfreies Überleben und teils auch ein besseres Gesamtüberleben als Patienten mit MRD-positiver kompletter Remission (13). Ausserhalb von klinischen Studien werden eine routinemässige MRD-Bestimmung wie auch Therapien zur MRDEradikation nicht empfohlen (12).
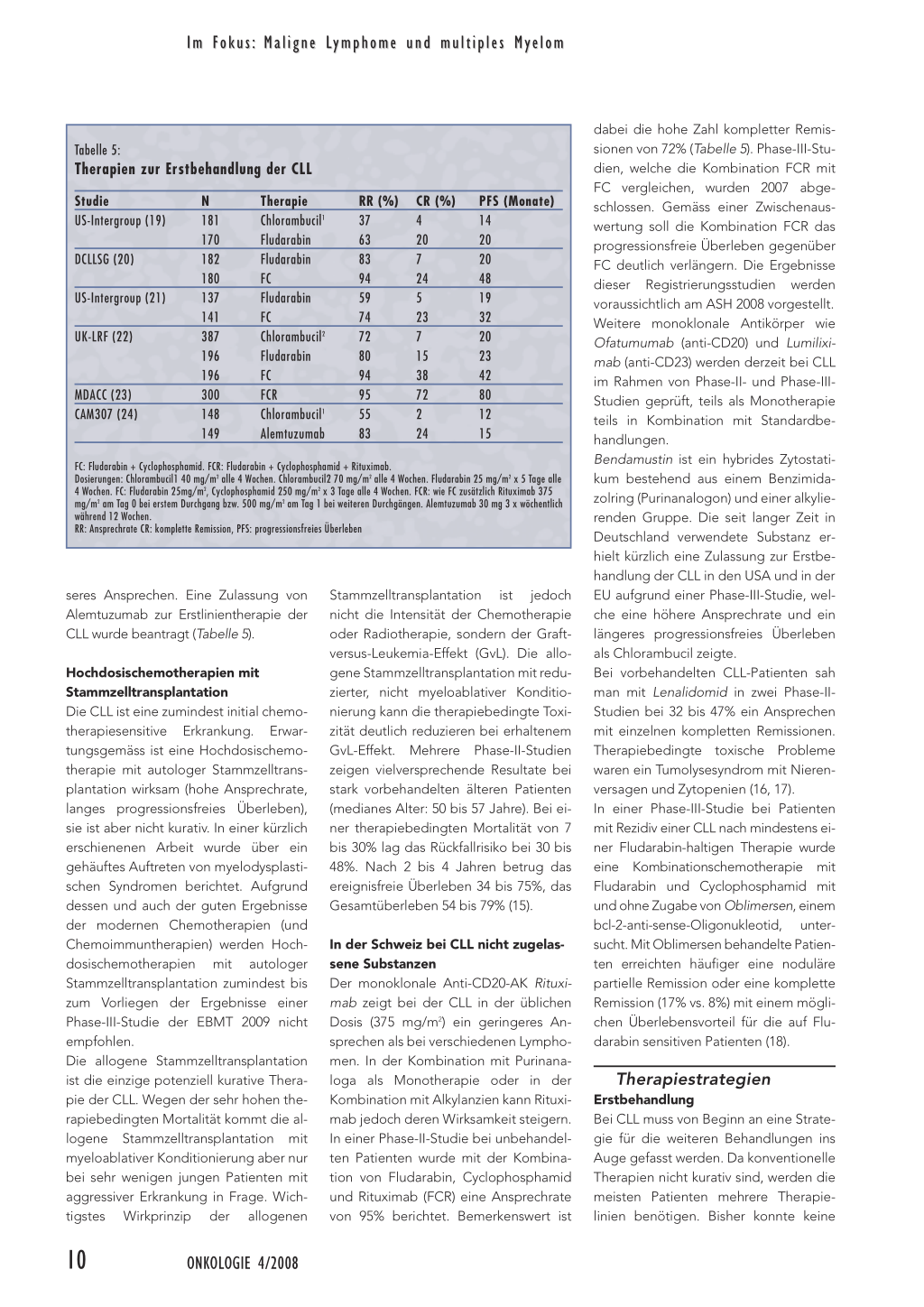
Chemotherapien Zahlreiche Chemotherapeutika sind bei CLL wirksam. In der folgenden Zusammenstellung werden die am häufigsten verwendeten Substanzen und Kombinationen diskutiert. Bei allen Studien ist zu beachten, dass Studienpatienten etwa 5 bis 10 Jahre jünger sind als der Median der CLL-Patienten und dass Patienten mit relevanten Komorbiditäten von der Studienteilnahme ausgeschlossen sind (Tabelle 5).
Chlorambucil Das Alkylans Chlorambucil war während mehreren Jahrzehnten der Standard zur Erstbehandlung der CLL. Chlorambucil kann oral eingenommen werden und hat nur wenige Nebenwirkungen (kaum Nausea, keine Alopezie, keine schweren Zytopenien). Es gibt zahlreiche Dosierungsschemata für Chlorambucil. Eine intermittierende Gabe über mehrere Tage wird gegenüber einer kontinuierlichen Therapie meist bevorzugt. Das Ansprechen auf Chlorambucil ist teilweise dosisabhängig. Kombinationschemotherapien (CAP, COP, CHOP) zeigten im Vergleich zu Chlorambucil allein eine höhere Ansprechrate, aber keinen Überlebensvorteil.
Tabelle 4:
Remissionskriterien nach IWCLL 2007 (11)
Komplette Remission (erfordert alle 4 Kriterien) ▲ keine konstitutionellen Symptome ▲ keine palpable Lymphadenopathie (> 1,5 cm im Durchmesser) oder Organomegalie ▲ Blutwerte1 : Neutrophile Granulozyten > 1,5 G/l, Thrombozyten > 100 G/l, Hämoglobin > 110 g/l ▲ kein Nachweis von klonalen Lymphozyten im Blut2
Partielle Remission (mindestens 1 Kriterium erforderlich) ▲ keine konstitutionellen Symptome ▲ Reduktion der Lymphozytose um > 50% ▲ Reduktion der Lymphadenopathie oder Organomegalie um > 50% ▲ Blutwerte1 : Neutrophile Granulozyten > 1,5 G/l, Thrombozyten > 100 G/l, Hämoglobin > 110 g/l
oder >50% Anstieg vom Ausgangswert.
Das Ansprechen sollte 3 Monate nach Therapieabschluss beurteilt werden. Für die Diagnose einer kompletten oder partiellen Remission ist eine Remissionsdauer von mindestens 2 Monaten erforderlich. Bei einer Remissionsdauer von unter 6 Monaten gilt die Erkrankung als refraktär auf die zuletzt verabreichte Therapie. 1 Ohne Transfusionen oder Wachstumsfaktoren. 2 mittel konventioneller Immunphänotypisierung (nicht MRD-Diagnostik!)
Purinanaloga Die Purinanaloga Flurarabin und Cladribin (und das in der Schweiz nicht zugelassene Pentostatin) zeigten in den ersten Phase-II- und Phase-III-Studien eine gegenüber Chlorambucil deutlich höhere Ansprechrate, eine bessere Remissionsqualität (mehr komplette Remissionen) und teilweise ein längeres progressionsfreies Überleben. Eine initial vermutete Verlängerung des Gesamtüberlebens konnte jedoch nicht nachgewiesen werden. Eine neuere Studie stellt auch die Verlängerung des progressionsfreien Überlebens in Frage (22). Dennoch galt eine Monotherapie mit Purinanaloga in vielen Ländern als Standardtherapie für die Erstbehandlung jüngerer Patienten ohne relevante Komorbidität (Kontraindikation: Niereninsuffizienz mit Clearance < 70ml/min !). Kombinationstherapien mit Purinanaloga In drei Studien wurde die Überlegenheit der Kombination Fludarabin und Cyclophosphamid (FC) gegenüber FludarabinMonotherapie bezüglich Ansprechrate, Remissionsqualität (mehr komplette Remissionen) und progressionsfreien Überlebens, jedoch nicht bezüglich Gesamtüberlebens nachgewiesen (20–22). Die Kombinationschemotherapie mit FC gilt heute als Chemotherapiestandard für Patienten < 65 bis 70 Jahren in gutem Allge- meinzustand ohne relevante Komorbidität. Die Kombinationstherapien hat eine höhere therapiebedingte Toxizität als eine Fludarabin-Monotherapie. Alemtuzumab Der gegen CD52 gerichtete monoklonale Antikörper Alemtuzumab wurde zur Behandlung der auf Purinanaloga refraktären CLL zugelassen (14). Bei Fludarabin-refraktären Patienten und bei Frührezidiven nach Kombinationschemotherapien mit Purinanaloga oder Immunchemotherapien kann mit Alemtuzumab bei etwa der Hälfte der Patienten ein erneutes Ansprechen erzielt werden. Bei ausgeprägter Lymphadenopathie ist eine Kombination von Alemtuzumab mit hochdosierten Steroiden angezeigt. Alemtuzumab ist auch bei Patienten mit der prognostisch ungünstigen del(17p) gut wirksam, bei welcher Alkylantien und Purinanaloga eine tiefe Ansprechrate zeigten. Infusionsbedingte Nebenwirkungen von Alemtuzumab sind bei subkutaner Gabe deutlich geringer. Wegen der therapiebedingten Immunsuppression und hohen Infektionsrate wird eine Prophylaxe gegen Pneumocystis jiroveci und Herpes-Viren empfohlen. Daneben ist eine Frühdiagnostik bezüglich Zytomegalovirus-Reaktivierung notwendig. Eine Studie zeigte in der Ersttherapie ein gegenüber Chlorambucil signifikant bes- ONKOLOGIE 4/2008 9 Im Fokus: Maligne Lymphome und multiples Myelom Tabelle 5: Therapien zur Erstbehandlung der CLL Studie US-Intergroup (19) DCLLSG (20) US-Intergroup (21) UK-LRF (22) MDACC (23) CAM307 (24) N 181 170 182 180 137 141 387 196 196 300 148 149 Therapie Chlorambucil1 Fludarabin Fludarabin FC Fludarabin FC Chlorambucil2 Fludarabin FC FCR Chlorambucil1 Alemtuzumab RR (%) 37 63 83 94 59 74 72 80 94 95 55 83 CR (%) 4 20 7 24 5 23 7 15 38 72 2 24 PFS (Monate) 14 20 20 48 19 32 20 23 42 80 12 15 FC: Fludarabin + Cyclophosphamid. FCR: Fludarabin + Cyclophosphamid + Rituximab. Dosierungen: Chlorambucil1 40 mg/m2 alle 4 Wochen. Chlorambucil2 70 mg/m2 alle 4 Wochen. Fludarabin 25 mg/m2 x 5 Tage alle 4 Wochen. FC: Fludarabin 25mg/m2, Cyclophosphamid 250 mg/m2 x 3 Tage alle 4 Wochen. FCR: wie FC zusätzlich Rituximab 375 mg/m2 am Tag 0 bei erstem Durchgang bzw. 500 mg/m2 am Tag 1 bei weiteren Durchgängen. Alemtuzumab 30 mg 3 x wöchentlich während 12 Wochen. RR: Ansprechrate CR: komplette Remission, PFS: progressionsfreies Überleben seres Ansprechen. Eine Zulassung von Alemtuzumab zur Erstlinientherapie der CLL wurde beantragt (Tabelle 5). Hochdosischemotherapien mit Stammzelltransplantation Die CLL ist eine zumindest initial chemotherapiesensitive Erkrankung. Erwartungsgemäss ist eine Hochdosischemotherapie mit autologer Stammzelltransplantation wirksam (hohe Ansprechrate, langes progressionsfreies Überleben), sie ist aber nicht kurativ. In einer kürzlich erschienenen Arbeit wurde über ein gehäuftes Auftreten von myelodysplastischen Syndromen berichtet. Aufgrund dessen und auch der guten Ergebnisse der modernen Chemotherapien (und Chemoimmuntherapien) werden Hochdosischemotherapien mit autologer Stammzelltransplantation zumindest bis zum Vorliegen der Ergebnisse einer Phase-III-Studie der EBMT 2009 nicht empfohlen. Die allogene Stammzelltransplantation ist die einzige potenziell kurative Therapie der CLL. Wegen der sehr hohen therapiebedingten Mortalität kommt die allogene Stammzelltransplantation mit myeloablativer Konditionierung aber nur bei sehr wenigen jungen Patienten mit aggressiver Erkrankung in Frage. Wichtigstes Wirkprinzip der allogenen Stammzelltransplantation ist jedoch nicht die Intensität der Chemotherapie oder Radiotherapie, sondern der Graftversus-Leukemia-Effekt (GvL). Die allogene Stammzelltransplantation mit reduzierter, nicht myeloablativer Konditionierung kann die therapiebedingte Toxizität deutlich reduzieren bei erhaltenem GvL-Effekt. Mehrere Phase-II-Studien zeigen vielversprechende Resultate bei stark vorbehandelten älteren Patienten (medianes Alter: 50 bis 57 Jahre). Bei einer therapiebedingten Mortalität von 7 bis 30% lag das Rückfallrisiko bei 30 bis 48%. Nach 2 bis 4 Jahren betrug das ereignisfreie Überleben 34 bis 75%, das Gesamtüberleben 54 bis 79% (15). In der Schweiz bei CLL nicht zugelassene Substanzen Der monoklonale Anti-CD20-AK Rituximab zeigt bei der CLL in der üblichen Dosis (375 mg/m2) ein geringeres Ansprechen als bei verschiedenen Lymphomen. In der Kombination mit Purinanaloga als Monotherapie oder in der Kombination mit Alkylanzien kann Rituximab jedoch deren Wirksamkeit steigern. In einer Phase-II-Studie bei unbehandelten Patienten wurde mit der Kombination von Fludarabin, Cyclophosphamid und Rituximab (FCR) eine Ansprechrate von 95% berichtet. Bemerkenswert ist dabei die hohe Zahl kompletter Remissionen von 72% (Tabelle 5). Phase-III-Studien, welche die Kombination FCR mit FC vergleichen, wurden 2007 abgeschlossen. Gemäss einer Zwischenauswertung soll die Kombination FCR das progressionsfreie Überleben gegenüber FC deutlich verlängern. Die Ergebnisse dieser Registrierungsstudien werden voraussichtlich am ASH 2008 vorgestellt. Weitere monoklonale Antikörper wie Ofatumumab (anti-CD20) und Lumiliximab (anti-CD23) werden derzeit bei CLL im Rahmen von Phase-II- und Phase-IIIStudien geprüft, teils als Monotherapie teils in Kombination mit Standardbehandlungen. Bendamustin ist ein hybrides Zytostatikum bestehend aus einem Benzimidazolring (Purinanalogon) und einer alkylierenden Gruppe. Die seit langer Zeit in Deutschland verwendete Substanz erhielt kürzlich eine Zulassung zur Erstbehandlung der CLL in den USA und in der EU aufgrund einer Phase-III-Studie, welche eine höhere Ansprechrate und ein längeres progressionsfreies Überleben als Chlorambucil zeigte. Bei vorbehandelten CLL-Patienten sah man mit Lenalidomid in zwei Phase-IIStudien bei 32 bis 47% ein Ansprechen mit einzelnen kompletten Remissionen. Therapiebedingte toxische Probleme waren ein Tumolysesyndrom mit Nierenversagen und Zytopenien (16, 17). In einer Phase-III-Studie bei Patienten mit Rezidiv einer CLL nach mindestens einer Fludarabin-haltigen Therapie wurde eine Kombinationschemotherapie mit Fludarabin und Cyclophosphamid mit und ohne Zugabe von Oblimersen, einem bcl-2-anti-sense-Oligonukleotid, untersucht. Mit Oblimersen behandelte Patienten erreichten häufiger eine noduläre partielle Remission oder eine komplette Remission (17% vs. 8%) mit einem möglichen Überlebensvorteil für die auf Fludarabin sensitiven Patienten (18). Therapiestrategien Erstbehandlung Bei CLL muss von Beginn an eine Strategie für die weiteren Behandlungen ins Auge gefasst werden. Da konventionelle Therapien nicht kurativ sind, werden die meisten Patienten mehrere Therapielinien benötigen. Bisher konnte keine 10 ONKOLOGIE 4/2008 Im Fokus: Maligne Lymphome und multiples Myelom Studie mit einer bestimmten Chemotherapiesequenz eine Verlängerung des Gesamtüberlebens zeigen. Dies ist durch die Wirkung weiterer Behandlungen und die Möglichkeit eines Cross-over bedingt. Die Entscheidung für eine bestimmte Therapie beruht heute vorwiegend auf Patientenfaktoren (Alter, Performance Score, Komorbidität). Der einzige prognostische Faktor, der die Therapiewahl heute beeinflusst, ist die Zytogenetik (FISH). Bei älteren Patienten (> 70 Jahre) oder bei relevanter Komorbidität ist Chlorambucil weiterhin Therapie der ersten Wahl. Bei jüngeren Patienten (< 70 Jahre) ohne Komorbidität sollte der primäre Einsatz einer intensiveren Therapie empfohlen werden. Das primäre Therapieziel ist dabei eine lange andauernde erste Remission. Der heutige Chemotherapiestandard in dieser Situation ist die Kombination Fludarabin und Cyclophosphamid. Aufgrund der vorliegenden Phase-II-Daten kann auch eine Chemoimmuntherapie mit Fludarabin, Cyclophosphamid und Rituximab als Ersttherapie verabreicht werden. Für eine generelle Empfehlung dieser Kombination sollten aber die Ergebnisse der abgeschlossenen Phase III-Studie abgewartet werden, welche voraussichtlich am ASH 2008 vorgestellt und 2009 publiziert werden. Bei allen Patienten mit del(17p) sollte bereits als Ersttherapie der Einsatz von Alemtuzumab erwogen werden, sei es als Monotherapie, sei es in Kombination mit hochdosierten Steroiden bei ausgeprägter Lymphadenopathie. Rezidivbehandlung Die Indikationen zur Einleitung einer Therapie im Rezidiv sind dieselben wie bei der Erstbehandlung. Die Wahl der Therapie hängt neben Patientenfaktoren auch von der Art der Primärtherapie und der Remissionsdauer ab. Bei längerer Remissionsdauer, je nach verabreichter Therapie 6 bis 24 Monate, kann prinzipiell dieselbe Therapie wiederholt werden. Die notwendige Remissionsdauer oder das therapiefreie Intervall, bei welchem das Wiedereinsetzen einer Therapie sinnvoll ist, hängt einerseits von der letzten Therapie, andererseits von den ver- Patienten < 65–70 Jahre PS ≤ 2, keine Komorbidität Patienten mit del(17p) Patienten > 65–70 Jahre PS > 2, mit Komorbidität
FC (+R)1
Alemtuzumab2
spät
spät
Rezidiv
Rezidiv
spät
früh
früh
Chlorambucil
Rezidiv
früh
Alemtuzumab3 Experimentelle
Therapie
Allogene SZT mit RIC bei geeigneten Patienten
Individuell: Fludarabin, Alemtuzumab, CVP
Experimentelle Therapie
Abbildung 2: Therapie der behandlungsbedürftigen CLL Eine Studienteilnahme sollte sowohl bei Erstbehandlung wie bei Rezidivbehandlung angestrebt werden!
Abkürzungen: PS: ECOG-Performancestatus, F: Fludarabin, C: Cyclophosphamid, R: Rituximab, SZT: Stammzelltransplantation. RIC: reduced intensity conditioning = nicht myeloablative Konditionierung. CVP: Cyclophosphamid, Vincristin, Prednison. 1 Rituximab kann bereits bei Erstbehandlung verabreicht werden, sofern es von der Krankenkasse übernommen wird.
Rituximab ist bei Rezidiv als Orphan Indication meist verfügbar und sollte bei Rezidiv nach FC eingesetzt werden. 2 In Kombination mit hochdosierten Steroiden, falls eine ausgeprägte Lymphadenopathie vorliegt (> 5cm).
Tabelle 6:
Indikationen für allogene Stammzelltransplantation bei CLL (nach 25)
1. Rezidive innert 12 Monaten nach Therapie mit Purinanaloga 2. Rezidive innert 24 Monaten nach Purinanaloga-enthaltenden Kombinationschemotherapien oder
autologer Stammzelltransplantation 3. Therapiebedürftige Patienten mit del(17p) oder p53-Mutation
bleibenden Therapieoptionen ab. Bei einem älteren polymorbiden Patienten mit acht Monaten therapiefreiem Intervall nach Chlorambucil kann das Wiedereinsetzen desselben Medikaments durchaus sinnvoll sein. Bei einem jüngeren, ausser der CLL gesunden Patienten, bei welchem mittels Chemoimmuntherapie mit Fludarabin, Cyclophosphamid und Rituximab eine relativ kurze Remissionsdauer mit einem therapiefreien Intervall von nur 20 Monaten erreicht wurde, sollten andere Therapieoptionen geprüft werden. Bei geringgradigem Ansprechen (partielle Remission oder schlechter) oder kürzerer Remissionsdauer wird empfohlen, die Therapie zu wechseln. Eine allogene Stammzelltransplantation sollte bei allen jüngeren Patienten (< 65 Jahre) mit Frührezidiv, schlechtem Ansprechen auf eine intensive Therapie oder sehr schlechter Prognose evaluiert werden (Tabelle 6). Konklusion Mittels prognostischer Marker kann heute der heterogene Verlauf der CLL abgeschätzt werden. Die intensiveren Kombinationschemotherapien und die Chemoimmuntherapie haben bei jünge- ren Patienten zu einer relevanten Verlän- gerung der therapiefreien Zeit geführt. Es ist dagegen heute unklar, ob MRD-ge- steuerte Strategien zu einer Verbesse- rung der Therapieergebnisse führen. Ein genaueres Verständnis der Pathogenese und der Risikofaktoren wird in Zukunft zu einer weiteren Verbesserung der CLL- Therapie führen. Ziel ist eine risikoadap- tierte Therapie, welche unter Berücksich- tigung von Patientenfaktoren und der individuellen Biologie der Erkrankung zu einer Langzeitkontrolle oder Eradikation der CLL führt. Dafür ist der Einschluss al- ler geeigneten Patienten in klinische Stu- dien anzustreben. ▲ ONKOLOGIE 4/2008 11 Im Fokus: Maligne Lymphome und multiples Myelom Dr. med. Michael Gregor Hämatologische Abteilung Departement Medizin Kantonsspital Luzern 6000 Luzern 16 E-Mail: michael.gregor@ksl.ch Quellen: 1. Ghia P: Hematology Education: the education program for the annual congress of the European Hematology Association. Abstract-Band 2008; 2: 302–7. 2. Rai KR et al.: Blood 1975; 46: 219–34. 3. Binet JL et al.: Cancer 1981; 48: 198–204. 4. Montserrat E et al.: Br J Haematol 1986; 62: 567– 75. 5. Hallek M et al.: Leuk Lymphoma 1996; 22: 439– 47. 6. Damle RN et al.: Blood 1999; 94: 1840–7. 7. Crespo M et al.: NEJM 2003; 348: 1764–75. 8. Hamblin TJ et al.: Blood 1999; 94: 1848–54. 9. Thorsélius M et al.: Blood 2006; 107: 2889–94. 10. Döhner H et al.: NEJM 2000; 343: 1910–6. 11. Lozanski G et al.: Blood 2004; 103: 3278–81. 12. Hallek M et al.: Blood 2008; 111: 5446–56. 13. Moreton P et al: J Clin Oncol 2005; 23: 2971–79. 14. Keating MJ et al.: Blood 2002; 99: 3554–61. 15. Dreger P: Hematology Education: the education program for the annual congress of the European Hematology Association. Abstract-Band 2008; 2: 314–19. 16. Ferrajoli A et al.: Blood 2008; 111: 5291–97. 17. Andritsos LA et al.: J Clin Oncol 2008; 26: 2519–25. 18. O’Brien S et al.: J Clin Oncol 2007; 25: 1114–20. 19. Rai K et al.: NEJM 2000; 343: 1750–57. 20. Eichhorst BF et al.: (for the German CLL Study Group): Blood 2006; 107: 885–91. 21. Flinn IW et al.: J Clin Oncol 2007; 25: 793–98. 22. Catovsky D et al.: (for the UK National Cancer Research Institute Haematological Oncology Clinical Studies Group and NCRI Chronic Lymphocytic Leukaemia Working Group): Lancet 2007; 370: 230–39. 23. Tam CS et al.: Blood 2008; prepublished online April 14 2008. 24. Hillmen P et al.: J Clin Oncol 2007; 25: 5616–23. 25. Dreger P et al.: Leukemia 2007; 21: 12–7.