


Transkript
Im Fokus: Hereditäre Tumorkrankheiten bei Frauen
Hereditäre Hauttumoren speziell der Frau
Charakteristika, Genetik und Therapie verschiedener Formen Hereditäre Tumorkrankheiten der Haut bilden nur einen kleinen Anteil der Hauttumoren. Wir stellen aus einer Vielfalt verschiedener erblicher Formen fünf unterschiedliche Erkrankungen vor, die ein spezielles Risiko für das weibliche Geschlecht aufweisen.
BETTINA BURGER, PETER ITIN
Trotz der relativen Seltenheit hereditärer Hauttumoren ist es wichtig, diese Formen näher zu studieren, denn durch das Verständnis dieser Erkrankungen lassen sich Erkenntnisse für die sporadischen Formen gewinnen (1). Erbliche Tumorkrankheiten der Haut stellen Modellerkrankungen für die Pathogenese von spontanen Hauttumoren dar und machen es möglich, die zugrunde liegenden Ursachen und die klinische Relevanz im menschlichen System zu untersuchen. Auf dieser Basis können Therapiekonzepte entwickelt werden, die Bedeutung für die Gesamtbevölkerung erlangen können.
Basalzellnävus-Syndrome
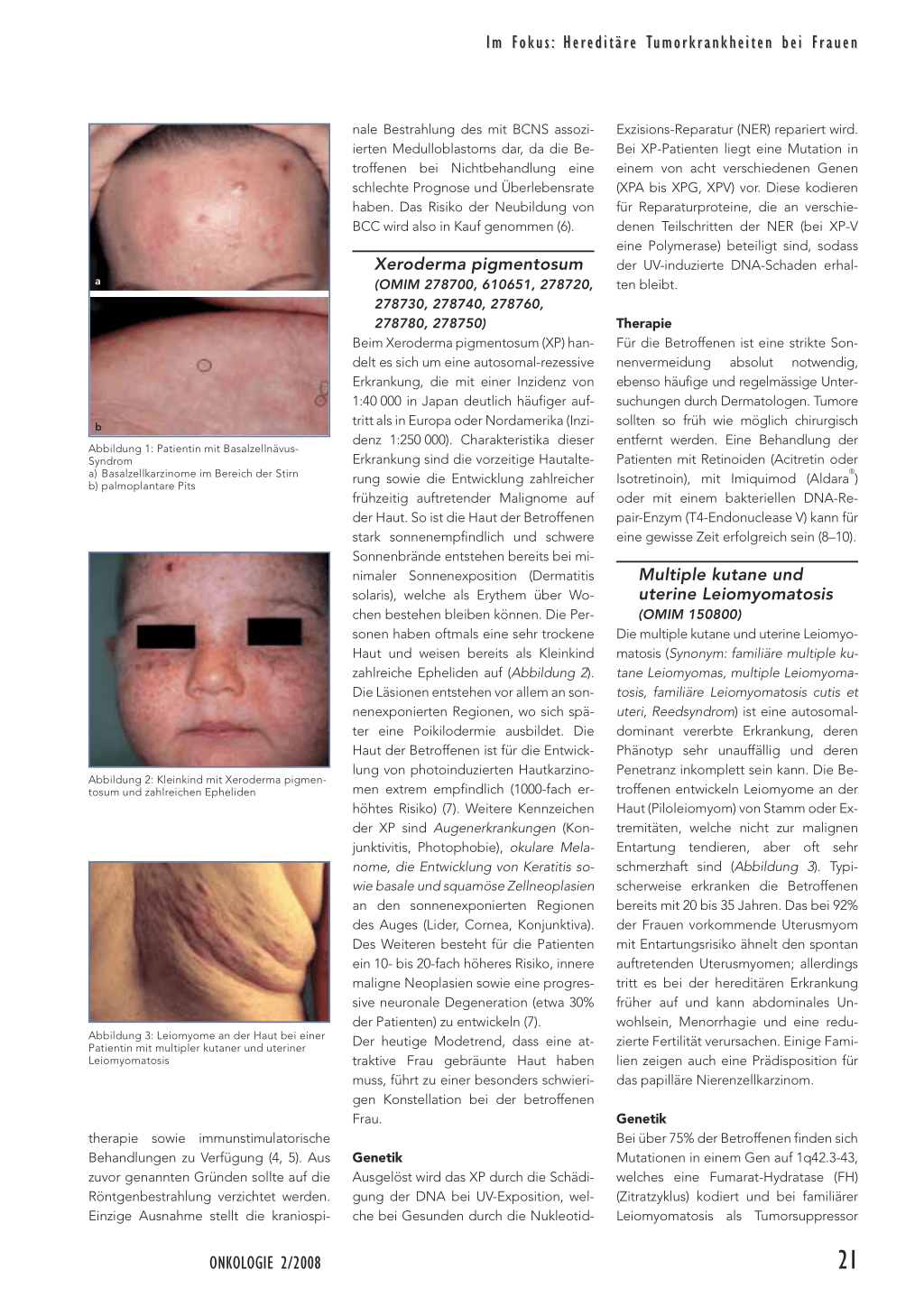
(OMIM 109400) Das Basalzellnävus-Syndrom (BCNS) (auch Gorlin Syndrom oder Nevoid basal cell carcinoma syndrome [NBCCS] genannt) ist eine autosomal-dominant vererbte Erkrankung mit einer Prävalenz von 1:60 000, einer hohen Penetranz und variabler Expression (1). Die Diagnose erfolgt anhand von Hauptkriterien: ▲ einige wenige bis tausend Basalzellkarzinome
(BCC) ▲ odontogene Kieferzysten ▲ palmoplantare Pits ▲ Verkalkung der Falx cerebri und/oder ▲ Verwandte 1. Grades mit BCNS von denen mindestens zwei vorliegen müssen, und Nebenkriterien: ▲ kongenitale Skelettanomalien ▲ kongenitale Fehlbildungen ▲ frontookzipetaler Kopfumfang über der 97. Per-
zentile ▲ Ovarial- und Kardialfibrome sowie das Medullo-
blastom
von welchen mindestens zwei gemeinsam mit einem Hauptkriterium auftreten müssen (Abbildung 1). Verschiedene Tumore sind mit der Erkrankung assoziiert, so das Fibro- und Rhabdomyosarkom sowie das Melanom. Bezüglich frauenspezifischer Risiken ist bedeutsam, dass die Entwicklung von Ovarialkarzinomen gehäuft gemeinsam mit dem BCNS auftritt. Die Diagnosestellung des BCNS wird unterstützt von der molekulargenetischen Diagnostik (s.u.). Durch die molekulare Bestätigung ist ein verbessertes klinisches Monitoring sowie die Wahl der besten Therapie möglich (z. B. Vermeidung der Radiotherapie zur Behandlung von Basalzellkarzinomen [BCC], da sich in bestrahlten Bereichen verstärkt erneut BCC bilden) (2).
Genetik Das für BCNS verantwortliche Gen wurde 1996 auf Chromosom 9q22.3 identifiziert. Es handelt sich dabei um das 34 kb grosse Tumorsuppressor-Gen PTCH. Kennzeichen eines Tumorsuppressor-Gens ist, dass die Betroffenen eine inaktivierende Keimbahnmutation des Gens tragen und erst dann einen Tumor entwickeln, wenn in dem verbleibenden Allel eine sporadische Mutation auftritt, oft induziert durch UV-Licht. Bei BCNS konnten bisher in den 23 Exons des Gens, welches für das Transmembranprotein PATCH kodiert, mehr als 60 inaktivierende Keimbahnmutationen beschrieben werden, die in der Folge zu einer Störung der Regulation von Zellzyklus und Apoptose führen (3). Eine Genotyp-PhänotypKorrelation konnte bisher nicht gezeigt werden.
Therapie Therapeutisch werden die mit BCNS assoziierten dermatologischen Veränderungen bisher hauptsächlich chirurgisch behandelt; als alternative oder zusätzliche Therapie stehen der CO2-Laser, die Kryo-
20 ONKOLOGIE 2/2008
Im Fokus: Hereditäre Tumorkrankheiten bei Frauen
a
b Abbildung 1: Patientin mit BasalzellnävusSyndrom a) Basalzellkarzinome im Bereich der Stirn b) palmoplantare Pits
Abbildung 2: Kleinkind mit Xeroderma pigmentosum und zahlreichen Epheliden
Abbildung 3: Leiomyome an der Haut bei einer Patientin mit multipler kutaner und uteriner Leiomyomatosis
therapie sowie immunstimulatorische Behandlungen zu Verfügung (4, 5). Aus zuvor genannten Gründen sollte auf die Röntgenbestrahlung verzichtet werden. Einzige Ausnahme stellt die kraniospi-
nale Bestrahlung des mit BCNS assoziierten Medulloblastoms dar, da die Betroffenen bei Nichtbehandlung eine schlechte Prognose und Überlebensrate haben. Das Risiko der Neubildung von BCC wird also in Kauf genommen (6).
Xeroderma pigmentosum
(OMIM 278700, 610651, 278720, 278730, 278740, 278760, 278780, 278750) Beim Xeroderma pigmentosum (XP) handelt es sich um eine autosomal-rezessive Erkrankung, die mit einer Inzidenz von 1:40 000 in Japan deutlich häufiger auftritt als in Europa oder Nordamerika (Inzidenz 1:250 000). Charakteristika dieser Erkrankung sind die vorzeitige Hautalterung sowie die Entwicklung zahlreicher frühzeitig auftretender Malignome auf der Haut. So ist die Haut der Betroffenen stark sonnenempfindlich und schwere Sonnenbrände entstehen bereits bei minimaler Sonnenexposition (Dermatitis solaris), welche als Erythem über Wochen bestehen bleiben können. Die Personen haben oftmals eine sehr trockene Haut und weisen bereits als Kleinkind zahlreiche Epheliden auf (Abbildung 2). Die Läsionen entstehen vor allem an sonnenexponierten Regionen, wo sich später eine Poikilodermie ausbildet. Die Haut der Betroffenen ist für die Entwicklung von photoinduzierten Hautkarzinomen extrem empfindlich (1000-fach erhöhtes Risiko) (7). Weitere Kennzeichen der XP sind Augenerkrankungen (Konjunktivitis, Photophobie), okulare Melanome, die Entwicklung von Keratitis sowie basale und squamöse Zellneoplasien an den sonnenexponierten Regionen des Auges (Lider, Cornea, Konjunktiva). Des Weiteren besteht für die Patienten ein 10- bis 20-fach höheres Risiko, innere maligne Neoplasien sowie eine progressive neuronale Degeneration (etwa 30% der Patienten) zu entwickeln (7). Der heutige Modetrend, dass eine attraktive Frau gebräunte Haut haben muss, führt zu einer besonders schwierigen Konstellation bei der betroffenen Frau.
Genetik Ausgelöst wird das XP durch die Schädigung der DNA bei UV-Exposition, welche bei Gesunden durch die Nukleotid-
Exzisions-Reparatur (NER) repariert wird. Bei XP-Patienten liegt eine Mutation in einem von acht verschiedenen Genen (XPA bis XPG, XPV) vor. Diese kodieren für Reparaturproteine, die an verschiedenen Teilschritten der NER (bei XP-V eine Polymerase) beteiligt sind, sodass der UV-induzierte DNA-Schaden erhalten bleibt.
Therapie Für die Betroffenen ist eine strikte Sonnenvermeidung absolut notwendig, ebenso häufige und regelmässige Untersuchungen durch Dermatologen. Tumore sollten so früh wie möglich chirurgisch entfernt werden. Eine Behandlung der Patienten mit Retinoiden (Acitretin oder Isotretinoin), mit Imiquimod (Aldara®) oder mit einem bakteriellen DNA-Repair-Enzym (T4-Endonuclease V) kann für eine gewisse Zeit erfolgreich sein (8–10).
Multiple kutane und uterine Leiomyomatosis
(OMIM 150800) Die multiple kutane und uterine Leiomyomatosis (Synonym: familiäre multiple kutane Leiomyomas, multiple Leiomyomatosis, familiäre Leiomyomatosis cutis et uteri, Reedsyndrom) ist eine autosomaldominant vererbte Erkrankung, deren Phänotyp sehr unauffällig und deren Penetranz inkomplett sein kann. Die Betroffenen entwickeln Leiomyome an der Haut (Piloleiomyom) von Stamm oder Extremitäten, welche nicht zur malignen Entartung tendieren, aber oft sehr schmerzhaft sind (Abbildung 3). Typischerweise erkranken die Betroffenen bereits mit 20 bis 35 Jahren. Das bei 92% der Frauen vorkommende Uterusmyom mit Entartungsrisiko ähnelt den spontan auftretenden Uterusmyomen; allerdings tritt es bei der hereditären Erkrankung früher auf und kann abdominales Unwohlsein, Menorrhagie und eine reduzierte Fertilität verursachen. Einige Familien zeigen auch eine Prädisposition für das papilläre Nierenzellkarzinom.
Genetik Bei über 75% der Betroffenen finden sich Mutationen in einem Gen auf 1q42.3-43, welches eine Fumarat-Hydratase (FH) (Zitratzyklus) kodiert und bei familiärer Leiomyomatosis als Tumorsuppressor
ONKOLOGIE 2/2008
21
Im Fokus: Hereditäre Tumorkrankheiten bei Frauen
wirkt (11). Dieser Mechanismus sowie die Assoziation der FH mit glatten Muskelzelltumoren ist noch nicht aufgeklärt.
Neurofibromatose Typ 1 und 2
(OMIM 162200 und 101000) Zu der Neurofibromatose (NF) sind acht verschiedene Typen beschrieben (12), von denen Typ 1 und Typ 2 die häufigsten sind. Bei beiden Formen handelt es sich um autosomal-dominante neurokutane Erkrankungen mit einer de-novo-Mutationsrate von mindestens 50%. Ebenfalls beiden Typen gemeinsam ist, dass ein Tumorsuppressor-Gen die Ursache der Erkrankung ist und eine komplette Penetranz aber variable Expression zeigt. Bei NF1 und NF2 handelt es sich klinisch und genetisch aber um unterschiedliche Erkrankungen. NF1 (auch Morbus Recklinghausen genannt) tritt mit einer Inzidenz von 1:3000 auf. Ihre Hauptmerkmale (13) sind ▲ neurokutane Fibrome ▲ Café-au-lait-Flecken ▲ Lisch-Knötchen und ▲ das sogenannte Freckling der Haut. NF2 (Synonym: bilaterale akustische oder zentrale Neurofibromatose) ist mit einer Prävalenz von 1:40 000 deutlich seltener als NF1 und zeigt nur wenige oder keine ihrer typischen Hautläsionen. Ihre Leitsymptome sind ▲ das bilaterale akutische Neurinom/
Schwannom (95%) ▲ multiple Meningiome ▲ Ependymome des Rückenmarks und ▲ Gliome sowie ▲ okulare Manifestationen (u.a. Kata-
rakt, retinale Hamartome). Eine neue Arbeit hat gezeigt, dass Progesteronrezeptoren bei der Entwicklung der Meningiome bedeutsam sind (14). Im Gegensatz zur NF1 verursachen bei einigen Patienten NF2-Neuropathien ein fortschreitendes neurologisches Defizit. Die kutanen Stigmata sind weniger auffällig als bei NF1, so treten Café-au-lait-Flecken weniger zahlreich (bei etwa 40% der Patienten) auf. Hauttumore entwickeln sich bei zwei Drittel der PatientInnen.
Genetik Bei NF1 ist das auf Chromosom 17q11.2 liegende Neurofibromin (bestehend aus
Tabelle:
Diagnostische Kriterien für die tuberöse Sklerose
Kutane Hauptkriterien ▲ hypomelanotische Maculae
(«Eschenlaubflecken») ▲ aziale Angiofibrome ▲ «shagreen patches» ▲ unguale Fibrome ohne Trauma
(Könen-Tumor)
Weitere Hauptkriterien ▲ kortikale Tubera ▲ retinale Hamartome ▲ kardiale Rhabdomyome ▲ Angiomyolipome der Niere ▲ pulmonare Lymphangioleiomyomatose ▲ subependymale Knoten ▲ subependymale Riesenzelltumore
Nebenkriterien ▲ Fibrome der Gingiva ▲ «konfettiähnliche» Hypopigmentierungen ▲ Nierenzysten ▲ multiple Pits am Zahnschmelz ▲ hamartomatöse rektale Polypen ▲ Knochenzysten ▲ retinale «achromic patches»
Häufigkeit und Alter bei Auftreten bei Geburt oder im 1. Lebensjahr (> 90%)
im Kleinkindalter (75%) oft während der Kindheit (48%) bei Jugendlichen oder Erwachsenen (15%)
Fötus (80%) Kleinkinder (50%) Fötus (50–70%) Kindheit bis Erwachsenenalter (55–75%) Jugend bis Erwachsenenalter (selten, aber schwer) Kindheit bis Jugend Kindheit bis Jugend
60 Exons und für 360 AS kodierend) von Mutationen betroffen (15). Diese können überall im Gen auftreten, wobei eine Genotyp-Phänotyp-Beziehung in den meisten Fällen unklar ist. Die Diagnose erfolgt klinisch, eine genetische Testung ist nur selten nötig, dennoch kann bei über 95% der Patienten die Mutation identifiziert werden. Mutationen im NF1-Gen resultieren im «loss of function» von Neurofibromin, was über einen Signalweg verschiedener Proteine zu einer erhöhten Proliferation und Tumorgenese führt. Das NF2 verursachende Gen liegt auf Chromosom 22q12.2 und kodiert in 17 Exons für das 595 AS grosse Protein Merlin (auch Schwannomin genannt). Merlin wirkt in multiplen interzellulären Signalwegen und verhindert das Zellwachstum. Auch hier ist in 95% der Fälle die Identifizierung der Mutation möglich.
Therapie Die Entwicklung von effektiven Behandlungen für Patienten mit NF1 oder NF2 steht noch aus.
Tuberöse Sklerose
(OMIM 191100) Diese autosomal-dominant vererbte neurokutane Multisystemerkrankung (auch hereditäre multiple Systemhamartomatosis oder tuberöser Sklerosekomplex genannt) tritt mit einer Prävalenz von 1:6000 bis 1:10 000 auf und ist durch tumorähnliche Läsionen (Hamartome) charakterisiert. Die Bezeichnung tuberöse Sklerose (TS) geht auf die Präsenz von multiplem sklerotisierendem Gewebe im Cerebrum zurück.
Diagnostik Da viele Stigmata erst während der späten Kindheit oder Jugend auftreten, ist die Diagnose der Erkrankung erschwert. Zudem weist die TS zwar eine komplette Penetranz, aber eine variable Expression auf, die auch innerhalb einer Familie sehr gross sein kann. Zu den kutanen Hauptkriterien zählen die hypomelanotischen Maculae («Eschenlaubflecken»), die bei über 90% aller PatientInnen vorliegen und bereits bei Geburt oder im ersten
22 ONKOLOGIE 2/2008
Im Fokus: Hereditäre Tumorkrankheiten bei Frauen
Lebensjahr auftreten. Im Kleinkindalter beziehungsweise während der Kindheit treten bei 75% die kennzeichnenden fazialen Angiofibrome (bzw. «Shagreen patches» 48%) auf, während die KönenTumore erst bei Jugendlichen oder Erwachsenen vorkommen (16). In einer prospektiven Studie wurde gezeigt, dass speziell Frauen mit TS gehäuft knotige und zystische Lungenveränderungen aufweisen (17). Eine Zusammenfassung aller Haupt- und Nebenkriterien zeigt die Tabelle. Zusätzlich treten häufig auch Café-au-lait-Flecken (30%) sowie fibröse Plaques an der Stirn auf. Viele Betroffene sind von Epilepsie (80%), mentaler Retardierung (44%) oder/und Autismus betroffen, 20 bis 30% der Kinder mit TS haben infantile Spasmen (18). Eine höhere Anzahl an Tubera (> 7) geht mit der Entwicklung infantiler Spasmen und schlecht behandelbarer Epilepsie einher, wobei die Region, welcher die Anfälle entspringen, mit der Lokalisation der Tubera im Gehirn koinzidiert. Daher wird eine anderweitig schlecht behandelbare Epilepsie oft mit der Resektion der Tubera behandelt. Die neurologischen Manifestationen weisen oftmals eine grosse Heterogenität auf und repräsentieren die führende Ursache für assoziierte Morbidität und Mortalität.
Genetik Bei 80% der PatientInnen kann in den für die Erkrankung verantwortlichen Genen eine Mutation identifiziert werden. Die pathogenetischen Sequenzveränderungen in den Genen TSC1 (kodiert für Hamartin) auf Chr 9q34 oder TSC2 (kodiert für Tuberin) auf Chromosom 16p13.3 sind nicht leicht festzustellen, da in beiden Genen ein grosser Anteil an Polymorphismen vorliegt. So zeigt TSC1 389 verschiedene allelische Varianten, von denen wenigstens 86 ohne erkennbaren Phänotypeffekt sind. TSC2 zeigt sogar 1107 verschiedene allelische Varianten, von welchen wenigstens 302 ohne erkennbaren Phänotypeffekt sind. Der aktuelle Stand der bekannten Varianten und ihres Phänotyps findet sich auf der Internetseite unter www.LOVD.nl/TSC1 (bzw. TSC2). Aufgrund der genetischen Variabilität sollte zurzeit die Diagnose klinisch laufen. Für eine verbesserte Diagnostik ist es notwendig, schneller und
verlässlicher pathogene Mutationen zu identifizieren. Da TSC1 und TSC2 ein Dimer bilden, wirken sich pathogene Mutationen auf denselben Signalweg aus. In der Folge fehlt die inhibitorische Wirkung des Hamartin/Tuberin-Komplexes, was zu einer erhöhten Aktivität von mTOR (= mammalian target of rapamycin) führt. Hierdurch kommt es nachfolgend zu einer Fehlregulation von Zellwachstum und Proliferation (19).
Therapie Bisher erfolgt eine Therapie hauptsächlich symptomatisch. Neue Möglichkeiten verspricht der Immunosuppressor Rapamycin (Sirolimus), welcher das dysregulierte mTOR inhibiert. In TSC1- und TSC2-Nullzelllinien sowie in Nagetiermodellen von TS konnte seine Wirkung bereits gezeigt werden, in ersten Behandlungen bei Menschen konnte ein Rückgang von Gehirnastrozytomen, assoziiert mit TS, induziert werden (20). ▲
Dr. rer. nat. Bettina Burger (Korrespondenzadresse) Departement Dermatologie und Departement Biomedizin Universitätsspital Basel Hebelstr. 20 4031 Basel E-Mail: burgerb@uhbs.ch
und
Prof. Dr. med. Peter Itin Chefarzt Dermatologische Klinik und Poliklinik Universitätsspital Basel Petersgraben 4 4031 Basel
1. High A, Zedan W: Basal cell nevus syndrome. Curr Opin Oncol 2005; 17: 160–166. 2. O’Malley S, Weitman D, et al.: Multiple neoplasms following craniospinal irradiation for medulloblastoma in a patient with nevoid basal cell carcinoma syndrome. Case report. J Neurosurg 1997; 86: 286–288. 3. Rubin LL, de Sauvage FJ: Targeting the hedgehog pathway in cancer. Nat Rev Drug Discov 2006; 5: 1026–1033. 4. Kopera D, Cerroni L, et al.: Different treatment modalities for the management of a patient with the nevoid basal cell carcinoma syndrome. J Am Acad Dermatol 1996; 34: 937–939. 5. Stockfleth E, Ulrich C, et al.: Successful treatment of basal cell carcinomas in a nevoid basal cell carcinoma syndrome with topical 5% imiquimod. Eur J Dermatol 2002; 12: 569–572.
6. Rupprecht M, Mensing CH, et al.: Skeletal and dermatological manifestations of the nevoid basal cell carcinoma syndrome. Results of 8 patients in 12 years. Rofo 2007; 179: 618–626.
7. Kraemer KH, Patronas NJ, et al.: Xeroderma pigmentosum, trichothiodystrophy and cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007; 145: 1388–1396.
8. Nagore E, Sevila A, et al.: Excellent response of basal cell carcinomas and pigmentary changes in Xeroderma pigmentosum to imiquimod 5% cream. Br J Dermatol 2003; 149: 858–861.
9. Yarosh D, Klein J, et al.: Effect of topically applied t4 endonuclease v in liposomes on skin cancer in Xeroderma pigmentosum: A randomised study. Xeroderma pigmentosum study group. Lancet 2001; 357: 926–929.
10. Marchetto MC, Correa RG, et al.: Functional lentiviral vectors for Xeroderma pigmentosum gene therapy. J Biotechnol 2006; 126: 424–430.
11. Tomlinson IP, Alam NA, et al.: Germline mutations in fh predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002; 30: 406–410.
12. Riccardi VM: Neurofibromatosis: Clinical heterogeneity. Curr Probl Cancer 1982; 7: 1–34.
13. Ferner RE: Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol 2007; 6: 340–351.
14. Claus EB, Park PJ, et al.: Specific genes expressed in association with progesterone receptors in meningioma. Cancer Res 2008; 68: 314–322.
15. Yohay KH: The genetic and molecular pathogenesis of nf1 and nf2. Semin Pediatr Neurol 2006; 13: 21–26.
16. Schwartz RA, Fernandez G, et al.: Tuberous sclerosis complex: Advances in diagnosis, genetics, and management. J Am Acad Dermatol 2007; 57: 189–202.
17. Franz DN, Brody A, et al.: Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. Am J Respir Crit Care Med 2001; 164: 661–668.
18. Joinson C, O’Callaghan FJ, et al.: Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med 2003; 33: 335–344.
19. Yates JR: Tuberous sclerosis. Eur J Hum Genet 2006; 14: 1065–1073.
20. Franz DN, et al.: Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 2006; 59: 490–498.
24 ONKOLOGIE 2/2008