



Transkript
Im Fokus: Hereditäre Tumorkrankheiten bei Frauen
Gynäkologische Malignome bei gastrointestinalen Tumorveranlagungen
Cowden-, Peutz-Jeghers- und Lynch-/HNPCC-Syndrom
Bei 5 bis 10% aller kolorektalen Karzinome liegt eine durchschlagskräftige, meist autosomaldominant vererbte Tumorveranlagung zugrunde. Auch wenn die gastroenterologische Vor- und Nachsorge der Anlageträgerinnen im Allgemeinen gut gelingt, wird oftmals übersehen, dass die meisten dieser Veranlagungen gleichsam zu verschiedenen anderen Krebsarten prädisponieren. Damit ist eine differenzierte, interdisziplinäre Betreuung dieser Frauen wesentlich.
KARL HEINIMANN, HANS F.A. VASEN
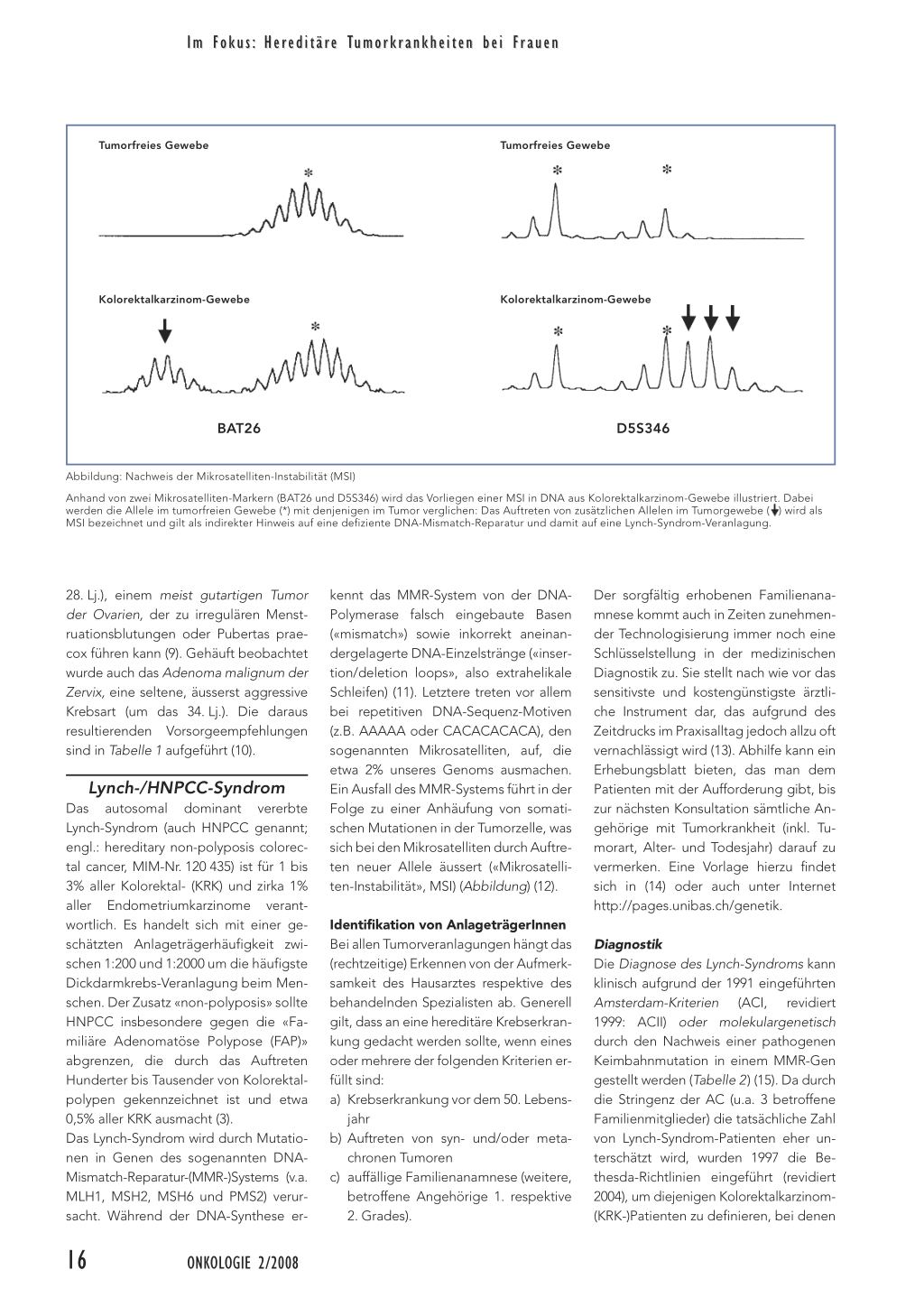
Mit der Entschlüsselung aller Gen-tragenden Abschnitte des menschlichen Genoms im Jahr 2003 wurde das viel zitierte «Jahrhundert der Biologie» eingeläutet (1). Die darauf aufbauende funktionelle Charakterisierung (Transcriptomics, Proteomics, Metabolomics) der 20 000 bis 25 000 Gene soll zu einem grundlegenden Verständnis von Gesundheit und Krankheit führen und so die Früherkennung, Therapie und Prävention krankheitsverursachender Prozesse ermöglichen. Das Zeitalter der «molekularen Medizin» darf jedoch nicht darüber hinwegtäuschen, dass bis dato nur von rund 400 Genen ihre klinische Bedeutung (Phänotyp) geklärt ist (2). Da die Mehrzahl dieser Veranlagungen zudem sehr selten (< 1/2500) sind, wird die entsprechende genetische Diagnostik meist nur von einigen wenigen (Forschungs-) Laboratorien in der Welt durchgeführt. Eine Ausnahme hierzu bilden die Veranlagungen zu gastrointestinalen Tumoren, die seit ihrer molekulargenetischen Charakterisierung in den Neunzigerjahren sowohl biologisch als auch klinisch eingehend untersucht worden sind (3). Die daraus gewonnenen Erkenntnisse konnten einerseits erfolgreich in die genetische Routinediagnostik umgesetzt werden. Andererseits ermöglichen sie konkrete, auf internationalem Konsens basierende Empfehlungen zur medizinischen Betreuung der Anlageträger und -trägerinnen. Hierbei hat sich auch deutlich gezeigt, dass sich die Vorsorge nicht nur auf ein Organ (z.B. den Gastrointestinaltrakt) beschränken darf, sondern dem spezifischen Spektrum der mit einer gegebenen Veranlagung assoziierten Tumorarten Rechnung tragen muss. Im Folgenden wird der Fokus auf die Diagnostik und Vorsorgemassnahmen bei denjenigen Kolorektaltumor-Veranlagungen gerichtet werden, die mit einem erhöhten Erkrankungsrisiko für Tumoren der weiblichen Geschlechtsorgane assoziiert sind. Neben den seltenen Hamartom-Polyposen, nämlich dem Cowden- und Peutz-Jeghers-Syndrom, die durch das Auftreten von hamartomatösen Gastrointestinalpolypen charakterisiert sind, soll schwergewichtig auf die häufigste Kolorektalkarzinom-Veranlagung, das LynchSyndrom (auch HNPCC, hereditary non-polyposis colorectal cancer genannt), eingegangen werden. Cowden-Syndrom Das autosomal dominant vererbte Cowden-Syndrom (MIM-Nr. 158350) (4) illustriert beispielhaft den Stellenwert der Molekulargenetik in der Klassifikation von (klinisch disparaten) Krankheitsveranlagungen. So wird das Cowden-Syndrom heute zusammen mit dem Bannayan-Riley-Ruvalcaba- (MIM-Nr. 153 480) und dem Proteus-Syndrom (MIM-Nr. 176 920) unter dem Begriff der «PTEN Hamartoma Tumor Syndrome» (PHTS) zusammengefasst: Allen diesen Veranlagungen liegen Keimbahnmutationen im 1997 identifizierten Tumorsuppressor-Gen PTEN (englisch: Phosphatase and tensin, deleted on chromosome TEN) auf dem langen Arm von Chromosom 10 zugrunde. Molekulargenetisch lässt sich bei zirka 80% aller Patienten mit Cowden-Syndrom eine pathogene PTENMutation nachweisen. Der durch PTEN kodierten «Dual-specificity-Phosphatase» kommt nach heutigem Verständnis eine zentrale Rolle in der Regulation des Zellzyklus (G1-Phase) sowie der Zellapoptose zu (5). Typische Begleiterkrankungen Diagnostisch wegweisend für das Cowden-Syndrom sind das Auftreten von mukokutanen Läsionen, insbesondere Trichilemmome und papillomatöse Ver- 14 ONKOLOGIE 2/2008 Im Fokus: Heriditäre Tumorkrankheiten bei Frauen Tabelle 1: Vorsorgeempfehlungen bei AnlageträgerInnen mit Cowden-, Peutz-Jeghersund Lynch-Syndrom (6, 10, 17) Tu m o r v e r a n l a g u n g Cowden-Syndrom Untere Altersgrenze (Jahre) 18 18 18 25 oder 5–10† 30–35 oder 5–10† 35–40 oder 5† 50 Peutz-Jeghers-Syndrom 8–10 10 25 20 30 Lynch-Syndrom 20–25 30–35 Familiäre Häufung von KRK ohne MSI 30–35 30–35 45–50 oder 5–10‡ Untersuchung Intervall (Jahre) Internistischer Status 1 Dermatologischer Status 1 Ultraschall Schilddrüse 1 Brustuntersuchung 1 Mammografie, 1 MRI der Brust Endometrium- 1 Aspirationsbiopsie Kolonoskopie (Baseline) 5* Obere und untere Endoskopie,2 Kapselendoskopie Hodenuntersuchung 1 Kolonoskopie 2 Brustuntersuch, 1 Mammografie, 2–3 PAP-Abstrich, 1 Endometrium- Aspirationsbiopsie Endoskopischer Ultraschall 1–2 (Pankreas), Abdominalultraschall Kolonoskopie 1–2 Transvaginaler Ultraschall, 1–2 Endometrium- Aspirationsbiopsie Gastroduodenoskopie** 1–2 Abdominalultraschall 1–2 Urinanalyse/-zytologie*** Kolonoskopie 3–5 † vor frühestem bekannten Brust- resp. Endometriumkarzinom in Familie ‡ vor frühestem bekannten Kolorektalkarzinom in Familie * Intervall abhängig vom Resultat ** falls Fälle von Magenkrebs in der Familie bekannt sind *** falls Fälle von Harnwegskrebs in der Familie bekannt sind Abkürzungen: KRK = Kolorektalkarzinom; MSI = Mikrosatelliten-Instabilität den-Syndrom sind in Tabelle 1 aufgeführt (6). Peutz-Jeghers-Syndrom Das Peutz-Jeghers-Syndrom (MIM-Nr. 175 200) ist charakterisiert durch das Auftreten von gastrointestinalen, hamartomatösen Polypen und einer peri-/enoral und perianal betonten Hyperpigmentierung. Die meist im Dünndarm lokalisierten Polypen können sich bereits im Kindesalter (10. bis 12. Lebensjahr) durch anämisierende chronische Blutungen und/oder kolikartige Bauchschmerzen infolge Invagination äussern. Der Nachweis von Peutz-Jeghers-Polypen, welche die histologisch typische verzweigte Verästelung der Muscularis mucosae aufweisen, gilt als diagnostisch wegweisend. Liegen gleichzeitig die charakteristischen «melanin spots» (v.a. Lippen- und Wangenschleimhaut, Finger), die allerdings in der Adoleszenz wieder abblassen können, und/oder eine positive Familiengeschichte (autosomal-dominanter Erbgang) vor, so sind formal die Kriterien eines Peutz-Jeghers-Syndroms erfüllt. Die Prävalenz der Veranlagung wird auf 1:25 000 bis 1:280 000 geschätzt. Molekulargenetisch kann bei nahezu 100% der Patienten mit positiver und bei etwa 90% derjenigen mit negativer Familiengeschichte eine pathogene Keimbahnmutation im STK11-Gen (Serin/Threonin-Protein-Kinase 11) auf dem kurzen Arm von Chromosom 19 nachgewiesen werden. Zu den tumorsupprimierenden Eigenschaften von STK11 zählen die Induktion der p53-vermittelten Apoptose sowie die Arretierung des Zellzyklus in der G1-Phase. änderungen im Gesichtsbereich sowie akrale Keratosen, die bis zum dritten Lebensjahrzehnt bei nahezu allen AnlageträgerInnen nachgewiesen werden können. Aufgrund der oftmals subtilen Hautmanifestationen wird die Prävalenz des Cowden-Syndroms mit 1:200 000 wohl eher unterschätzt. Neben dem Auftreten von gastrointestinalen, meist asymptomatischen Hamartomen (ca. 40%) werden bei etwa 75% der Patienten ein gutartiger Tumor der Schilddrüse (Struma, Adenom) und bei zirka 10% ein meist follikuläres Schilddrüsenkarzinom beobachtet. Rund 70% der betroffenen Frauen entwickeln Fibroadenome respektive zystische Veränderungen der Brust, und bei 25 bis 50% wird im Mittel um das 42. Lebensjahr ein Adenokarzinom der Brust diagnostiziert. Oft finden sich bei Patientinnen auch gutartige uterine Leiomyome (ca. 44%). Für ein Endometriumkarzinom wird ein Lebenszeitrisiko von 5 bis 10% angenommen. Die daraus abgeleiteten Vorsorgeempfehlungen für Anlageträgerinnen mit Cow- Das assoziiierte Tumorspektrum Studien an grösseren Kollektiven mit Peutz-Jeghers-Veranlagung haben ein kumulatives Krebsrisiko von 81 bis 93% bis zum 70. Lebensjahr (Lj.) errechnet (7, 8). Neben Tumoren des Magens (im Mittel um das 30. Lj.), des Pankreas (ca. 41. Lj.) und des Kolorektums (ca. 46. Lj.) zählen auch Östrogen-sezernierende Sertoli-Zell-Hodentumoren (10. Lj.) zum Tumorspektrum. Frauen mit PeutzJeghers-Syndrom tragen ein Risiko von 32 bis 54% für Brustkrebs (ca. 37. Lj.) sowie von zirka 21% für Keimstrangtumoren mit annulären Tubuli (SCTAT; ca. ONKOLOGIE 2/2008 15 Im Fokus: Hereditäre Tumorkrankheiten bei Frauen Tumorfreies Gewebe Tumorfreies Gewebe Kolorektalkarzinom-Gewebe Kolorektalkarzinom-Gewebe BAT26 D5S346 Abbildung: Nachweis der Mikrosatelliten-Instabilität (MSI) Anhand von zwei Mikrosatelliten-Markern (BAT26 und D5S346) wird das Vorliegen einer MSI in DNA aus Kolorektalkarzinom-Gewebe illustriert. Dabei werden die Allele im tumorfreien Gewebe (*) mit denjenigen im Tumor verglichen: Das Auftreten von zusätzlichen Allelen im Tumorgewebe ( ) wird als MSI bezeichnet und gilt als indirekter Hinweis auf eine defiziente DNA-Mismatch-Reparatur und damit auf eine Lynch-Syndrom-Veranlagung. 28. Lj.), einem meist gutartigen Tumor der Ovarien, der zu irregulären Menstruationsblutungen oder Pubertas praecox führen kann (9). Gehäuft beobachtet wurde auch das Adenoma malignum der Zervix, eine seltene, äusserst aggressive Krebsart (um das 34. Lj.). Die daraus resultierenden Vorsorgeempfehlungen sind in Tabelle 1 aufgeführt (10). Lynch-/HNPCC-Syndrom Das autosomal dominant vererbte Lynch-Syndrom (auch HNPCC genannt; engl.: hereditary non-polyposis colorectal cancer, MIM-Nr. 120 435) ist für 1 bis 3% aller Kolorektal- (KRK) und zirka 1% aller Endometriumkarzinome verantwortlich. Es handelt sich mit einer geschätzten Anlageträgerhäufigkeit zwischen 1:200 und 1:2000 um die häufigste Dickdarmkrebs-Veranlagung beim Menschen. Der Zusatz «non-polyposis» sollte HNPCC insbesondere gegen die «Familiäre Adenomatöse Polypose (FAP)» abgrenzen, die durch das Auftreten Hunderter bis Tausender von Kolorektalpolypen gekennzeichnet ist und etwa 0,5% aller KRK ausmacht (3). Das Lynch-Syndrom wird durch Mutationen in Genen des sogenannten DNAMismatch-Reparatur-(MMR-)Systems (v.a. MLH1, MSH2, MSH6 und PMS2) verursacht. Während der DNA-Synthese er- kennt das MMR-System von der DNAPolymerase falsch eingebaute Basen («mismatch») sowie inkorrekt aneinandergelagerte DNA-Einzelstränge («insertion/deletion loops», also extrahelikale Schleifen) (11). Letztere treten vor allem bei repetitiven DNA-Sequenz-Motiven (z.B. AAAAA oder CACACACACA), den sogenannten Mikrosatelliten, auf, die etwa 2% unseres Genoms ausmachen. Ein Ausfall des MMR-Systems führt in der Folge zu einer Anhäufung von somatischen Mutationen in der Tumorzelle, was sich bei den Mikrosatelliten durch Auftreten neuer Allele äussert («Mikrosatelliten-Instabilität», MSI) (Abbildung) (12). Identifikation von AnlageträgerInnen Bei allen Tumorveranlagungen hängt das (rechtzeitige) Erkennen von der Aufmerksamkeit des Hausarztes respektive des behandelnden Spezialisten ab. Generell gilt, dass an eine hereditäre Krebserkrankung gedacht werden sollte, wenn eines oder mehrere der folgenden Kriterien erfüllt sind: a) Krebserkrankung vor dem 50. Lebens- jahr b) Auftreten von syn- und/oder meta- chronen Tumoren c) auffällige Familienanamnese (weitere, betroffene Angehörige 1. respektive 2. Grades). Der sorgfältig erhobenen Familienanamnese kommt auch in Zeiten zunehmender Technologisierung immer noch eine Schlüsselstellung in der medizinischen Diagnostik zu. Sie stellt nach wie vor das sensitivste und kostengünstigste ärztliche Instrument dar, das aufgrund des Zeitdrucks im Praxisalltag jedoch allzu oft vernachlässigt wird (13). Abhilfe kann ein Erhebungsblatt bieten, das man dem Patienten mit der Aufforderung gibt, bis zur nächsten Konsultation sämtliche Angehörige mit Tumorkrankheit (inkl. Tumorart, Alter- und Todesjahr) darauf zu vermerken. Eine Vorlage hierzu findet sich in (14) oder auch unter Internet http://pages.unibas.ch/genetik. Diagnostik Die Diagnose des Lynch-Syndroms kann klinisch aufgrund der 1991 eingeführten Amsterdam-Kriterien (ACI, revidiert 1999: ACII) oder molekulargenetisch durch den Nachweis einer pathogenen Keimbahnmutation in einem MMR-Gen gestellt werden (Tabelle 2) (15). Da durch die Stringenz der AC (u.a. 3 betroffene Familienmitglieder) die tatsächliche Zahl von Lynch-Syndrom-Patienten eher unterschätzt wird, wurden 1997 die Bethesda-Richtlinien eingeführt (revidiert 2004), um diejenigen Kolorektalkarzinom(KRK-)Patienten zu definieren, bei denen 16 ONKOLOGIE 2/2008 Im Fokus: Hereditäre Tumorkrankheiten bei Frauen Tabelle 2: Amsterdam-Kriterien und revidierte Bethesda-Richtlinien (15, 16) ▲ Amsterdam-Kriterien I (ACI, 1991) Mindestens drei Familienangehörige mit KRK, wobei die folgenden Kriterien erfüllt sein müssen: 1. Eine(r) ist Verwandte(r) 1. Grades der beiden anderen. 2. Mindestens zwei aufeinanderfolgende Generationen sind betroffen. 3. Mindestens ein KRK wurde vor dem 50. Lebensjahr diagnostiziert. 4. Eine FAP ist bei den KRK-Patienten ausgeschlossen. 5. Die Tumoren wurden histopathologisch verifiziert. ▲ Amsterdam-Kriterien II (ACII, 1999) Mindestens drei Familienangehörige mit KRK oder einem Lynch-Syndrom-assoziierten Krebs (Endometrium, Dünndarm, Ureter oder Nierenbecken), wobei die folgenden Kriterien erfüllt sein müssen: 1. Eine(r) ist Verwandte(r) 1. Grades der beiden anderen. 2. Mindestens zwei aufeinanderfolgende Generationen sind betroffen. 3. Mindestens ein KRK wurde vor dem 50. Lebensjahr diagnostiziert. 4. Eine FAP ist bei den KRK-Patienten ausgeschlossen. 5. Die Tumoren wurden histopathologisch verifiziert. ▲ Revidierte Bethesda-Richtlinien (2004) Nur eines der folgenden Kriterien muss erfüllt sein: 1. Individuum, bei dem der KRK vor dem 50. Lebensjahr diagnostiziert wurde. 2. Auftreten syn- oder metachroner KRK oder anderer Lynch-Syndrom-assoziierter Tumoren (Magen, Dünndarm, Blase, Ureter, Nierenbecken, Gallenwege, Ovar, Pankreas, Hirn, TalgdrüsenAdenom und Keratoakanthome). 3. KRK mit MSI-typischer Histologie (Vorliegen von tumorinfiltrierenden Lymphozyten, Crohn’slike lymphozytische Reaktion, muzinöse/Siegelring-Differenzierung, medulläres Wachstumsmuster), die in einem Patienten vor dem 60. Lebensjahr diagnostiziert wurde. 4. Patient mit KRK und einem erstgradig Verwandten mit einem Lynch-Syndrom-assoziierten Tumor, wobei eine Krebserkrankung vor dem 50. Lebensjahr diagnostiziert wurde. 5. Patient mit KRK mit zwei oder mehr erst- oder zweitgradig Verwandten mit einem Lynch-Syndrom-assoziierten Tumor, unabhängig vom Alter bei Diagnosestellung. Abkürzungen: FAP = familiäre adenomatöse Polypose; KRK = Kolorektalkarzinom; MSI = Mikrosatelliten-Instabilität eine MSI-Abklärung durchgeführt werden soll (Tabelle 2) (16). Sind bei einem Patienten die ACII- und/oder die revidierten Bethesda-Richtlinien erfüllt, so sollte nach entsprechender genetischer Beratung und Aufklärung des Betroffenen eine molekularbiologische Abklärung eingeleitet werden. An dieser Stelle sei auf das per 1. April 2007 eingeführte Gesetz über genetische Untersuchungen beim Menschen (GUMG) hingewiesen, das insbesondere die Pflicht zur Information und Beratung der Betroffenen sowie der entsprechenden Dokumentation regelt (www.bag.admin.ch/genetictesting). In einem ersten Schritt wird molekulargenetisch das Vorliegen einer MSI und/ oder immunhistochemisch der Verlust von MMR-Proteinen (MLH1, MSH2, MSH6 und PMS2) im Tumorgewebe überprüft (12). Bei pathologischem Resultat wird in der Folge aus Leukozyten des Patienten isolierter DNA das entsprechende MMR-Gen auf Keimbahnmutationen hin überprüft. Abhängig von den Selektionskriterien und molekularbiologischen Befunden kann bei 8 bis 80% der Patienten eine pathogene Mutation in MLH1 oder MSH2 identifiziert werden. Der Nachweis einer Keimbahnmutation bei einem Betroffenen gibt dann weiteren Familienmitgliedern die Möglichkeit, ihren Trägerstatus bestimmen zu lassen (A-priori-Träger-Wahrscheinlichkeit 50%). Da es sich hierbei um eine präsymptomatische, also prädiktive Untersuchung handelt, darf diese jedoch nur nach adäquater genetischer Bera- tung – in der nicht nur die medizinischen, sondern auch die psychologischen und sozioökonomischen Aspekte (Versicherung, Beruf) eingehend besprochen werden – und mit der formalen Zustimmung des Ratsuchenden durchgeführt werden. Überwachung von AnlageträgerInnen Die Penetranz (Durchschlagskraft) des Lynch-Syndroms ist inkomplett. So wird geschätzt, dass 28 bis 75% aller männlichen und 24 bis 52% aller weiblichen Anlageträger während ihres Lebens an einem KRK erkranken werden (17). Weitere, gegenüber der Allgemeinbevölkerung gehäuft auftretende Tumoren betreffen insbesondere den Magen (2 bis 13%), den Dünndarm (4 bis 7%), die ableitenden Harnwege (1 bis 12%) und das hepatobiliäre System (ca. 2%). Anlageträgerinnen haben ein Lebenzeitrisiko von 27 bis 71% für ein Endometrium- und von 3 bis 13% für ein Ovarialkarzinom. Im Vergleich zur Kolonoskopie gibt es bis anhin nur wenige Studien zu Art (möglich: transvaginaler Ultraschall und/oder Aspirationsbiopsie), Intervall und Nutzen der gynäkologischen Überwachung beim Lynch-Syndrom: zwei von drei Studien identifizierten prämaligne Läsionen (komplexe Atypien in 3% resp. 7%), und eine Arbeit wies darauf hin, dass das Endometriumkarzinom damit in einem Frühstadium (Figo I) entdeckt werden kann (18–20). Die empfohlenen Vorsorge- und Überwachungsmassnahmen sind in Tabelle 1 aufgeführt. Bei abgeschlossener Familienplanung kann mit den Anlageträgerinnen die Option einer prophylaktischen Hysterektomie und bilateralen SalpingoOophorektomie erwogen werden. Schlussfolgerungen Die medizinische Betreuung von Anlage- trägerInnen hereditärer Tumorkrankhei- ten bedingt ein gut funktionierendes, meist durch den Hausarzt gesteuertes multidisziplinäres Netzwerk von Spezia- listen, namentlich der Chirurgie, Gastro- enterologie, Gynäkologie, medizinischen Genetik und Onkologie, damit das Wis- sen um die Veranlagung zum Vorteil der Betroffenen und deren Familien genutzt werden kann. ▲ ONKOLOGIE 2/2008 17 Im Fokus: Hereditäre Tumorkrankheiten bei Frauen PD Dr. med. et phil. II Karl Heinimann Abteilung Medizinische Genetik UKBB Römergasse 8 4005 Basel und Forschungsgruppe Humangenetik Zentrum für Biomedizin DBM Mattenstrasse 28 4058 Basel E-Mail: karl.heinimann@unibas.ch Dr. med. et phil. II Hans F.A. Vasen Department of Gastroenterology Leiden University Medical Centre und The Netherlands Foundation for the Detection of Hereditary Tumours Rijnsburgerweg 10 NL-2333 AA Leiden E-Mail: hfavasen@stoet.nl Quellen: 1. HumanGenomeProject: In: www.ornl.gov/sci/ techresources/Human_Genome/project/about.sht ml. 2. OMIM-Statistik: In: www.ncbi.nlm.nih.gov/ Omim/mimstats.html. 3. Lynch HT, de la Chapelle A: Hereditary colorectal cancer. N Engl J Med 2003; 348: 919–932. 4. OMIM-Datenbank: In: www.ncbi.nlm.nih.gov/ sites/entrez?db=omim. 5. Eng C: PTEN: one gene, many syndromes. Hum Mutat 2003; 22: 183–198. 6. Eng C: Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet 2000; 37: 828–830. 7. Giardiello FM, Brensinger JD, et al.: Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119: 1447–1453. 8. Lim W, Hearle N, et al.: Further observations on LKB1/STK11 status and cancer risk in PeutzJeghers syndrome. Br J Cancer 2003; 89: 308–313. 9. Hearle N, Schumacher V, et al.: Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006; 12: 3209–3215. 10. McGarrity TJ, Amos C: Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci 2006; 63: 2135–2144. 11. Jiricny J: The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 2006; 7: 335–346. 12. Heinimann K: Molekulargenetische Diagnostik bei HNPCC (Hereditäres Kolorektal-Karzinom ohne generalisierte Polypose). Schweiz Aerztezeitung 2000; 81: 2009–2012. 13. van Dijk DA, Oostindier MJ, et al.: Family history is neglected in the work-up of patients with colorectal cancer: a quality assessment using cancer registry data. Fam Cancer 2007; 6: 131–134. 14. Müller H, Heinimann K: Kolorektale Polypen – Wann sind Gentests angezeigt? Schweiz Med Forum 2002; 4: 59–66. 15. Vasen HF, Watson P, et al.: New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116: 1453–1456. 16. Umar A, Boland CR, et al.: Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96: 261–268. 17. Vasen HF, Moslein G, et al.: Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44: 353–362. 18. Dove-Edwin I, Boks D, et al.: The outcome of endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma. Cancer 2002; 94: 1708–1712. 19. Rijcken FE, Mourits MJ, et al.: Gynecologic screening in hereditary nonpolyposis colorectal cancer. Gynecol Oncol 2003; 91: 74–80. 20. Renkonen-Sinisalo L, Butzow R, et al.: Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int J Cancer 2007; 120: 821–824. 18 ONKOLOGIE 2/2008 Im Fokus: Hereditäre Tumorkrankheiten bei Frauen Diskussion: Konsequenzen für die Praxis Im Anschluss an das Referat von PD Dr. med. Karl Heinimann (Seite 14 bis 18) anlässlich der Fortbildung «Hereditäre Tumorkrankheiten der Frau» im Oktober 2007 diskutierte eine Expertengruppe aus den Disziplinen Medizinische Genetik, Gastroenterologie und Medizinische Onkologie die aktuellen Daten und Folgerungen für die medizinische Praxis bei Frauen mit Veranlagungen für Kolorektalkarzinome (KRK). HANSJAKOB MÜLLER (Resümee der Diskussion) Bedeutung der Familienanamnese Die Familienanamnese ist ein einfaches und kostengünstiges Mittel, um einer Krankheitsveranlagung auf die Spur zu kommen. Wenn gleiche oder assoziiert auftretende Tumorkrankheiten bei Angehörigen vorkommen, so sind diese für die Diagnosestellung praktisch gleichbedeutend wie diejenigen der Indexperson. Das Aufzeichnen eines krankheitsorientierten Stammbaums braucht Zeit. Die Ratsuchenden sollten sich entsprechend vorbereiten. Wir stellen ihnen dazu ein Schema im Internet zur Verfügung: http://pages.unibas. ch/genetik. → Diagnostik → Dokumente für HNPCC oder Hamartomatome. Umgang mit einem negativen Resultat des Gentests Gentests sind in der Regel zuverlässig, wenn sie durch erfahrenes Laborpersonal durchgeführt werden; ihre Ergebnisse sind in diesem Fall reproduzierbar. Ein negatives Resultat bedeutet, dass man keine Mutation in dem analysierten Gen fand. Biologische und auch technische Gründe können dafür verantwortlich sein. Vielleicht wurde das falsche Gen analysiert, oder es wurden Abschnitte im Erbgut nicht erfasst, die für dessen Regulation wichtig sind. Beim Peutz-Jeghers-Syndrom (PJS) findet man bei einer belasteten Familienanamnese in fast 100% der Patientinnen eine Mutation des STK11-Gens, jedoch in nur 90%, wenn keine solche vorliegt. In etwa 10% der Patientinnen mit Cowden-Syndrom (CS) kommt die Mutation nicht im kodierenden Abschnitt des PTEN-Gens Diskussionspartner: ● PD Dr. med. Rémy F. Meier, Gastroenterologie, Kantonsspital Liestal* ● PD Dr. med. Andreas N. Lohri, Onkologie, Kantonsspital Liestal* ● Prof. Dr. med. Hansjakob Müller Medizinische Genetik, Universität Basel** ● PD Dr. med. et phil. II Karl Heinimann, Medizinische Genetik, Universität Basel** * Leitende Ärzte, Kantonsspital Liestal ** Leitende Ärzte, Abtl. Medizinische Genetik, Universitäts-Kinderspital Beider Basel (UKBB) und Departement Biomedizin vor, sondern in dessen Promotorregion. Die Chance, Mutationen in einem der MMR-Gene, die zu HNPCC (Lynch-Syndrom) prädisponieren, zu finden, hängt von den klinisch-genealogischen Kriterien ab, die man anwendet. So haben die Amsterdam-Kriterien eine Sensitivität und Spezifität zwischen 60 und 70%. Mit den Bethesda-Kriterien steigt die Sensitivität über 90%, während dafür die Spezifität auf etwa 25% absinkt. Gemäss breiter Übereinstimmung sollte man einen Gentest in einer Familie vorerst bei einer erkrankten Person vornehmen, von der anzunehmen ist, dass sie die vermutete Mutation aufweist, und nicht bei einer gesunden Person, die ohnehin nur ein Risiko von 50% hat, die Genmutation geerbt zu haben. Kosten für molekulargenetische Abklärungen Gentests gelten allgemein als teuer. Dem ist nicht so! Ohne klare Indikation dürfen sie aber nicht veranlasst werden. Zu solchen zählen: ▲ klinische Symptome des entsprechen- den Syndroms ▲ ein frühes Erkrankungsalter ▲ das synchrone oder metachrone Auf- treten mehrerer Tumorkrankheiten bei der gleichen Person ▲ die belastete Familienanamnese sowie ▲ Eigenschaften des Tumors (Histologie, Mikrosatelliten-DNA-Instabilität, Immunhistochemie). Gemäss Analyseliste der KrankenpflegeLeistungsverordnung gelten DNA-Analysen bei klarem Verdacht auf ein relevantes molekulargenetisch bedingtes Leiden als Pflichtleistung. Gesetzliche Auflagen für genetische Untersuchungen Gemäss dem am 1. April 2007 in Kraft gesetzten Bundesgesetz über genetische Untersuchungen beim Menschen (GUMG) dürfen genetische Untersuchungen nur mit der Zustimmung der betroffenen Person nach hinreichender Aufklärung (Art. 6) durchgeführt werden. Ausschliesslich Ärztinnen und Ärzte können solche gemäss den Vorgaben in Artikel 13 veranlassen. Präsymptomatische Untersuchungen (Gentests bei gesunden Risikopersonen) müssen vor und nach ihrer Durchführung von einer nicht direktiven, fachkundigen genetischen Beratung begleitet werden, wobei das Beratungsgespräch zu dokumentieren ist (Art. 14). Gemäss Artikel 4 darf niemand wegen seines Erbguts diskriminiert werden. Im Versicherungsbereich besteht ein Nachforschungsverbot für Ergebnisse von Gentests (Art. 27), das bis zu einer Versicherungssumme von 400 000 Franken bei Lebensversicherungen und für freiwillige Invaliditätsversicherungen mit einer Jahresrente von höchstens 40 000 Franken gilt. Prof. Dr. med. Hansjakob Müller (Korrespondenzadresse) Abt. Medizinische Genetik UKBB/Dept. Biomedizin Römergasse 8 4005 Basel E-Mail: hansjakob.mueller@unibas.ch ONKOLOGIE 2/2008 19