



Transkript
SCHWERPUNKT
Hochrisikokonstellation für Brustkrebs
Empfehlungen zur genetischen Beratung und zur BRCA-Testung
Der «Angelina-Effekt»* – die prophylaktische Mastektomie bei der Schauspielerin Angelina Jolie – hat zu einer grossen Sensibilisierung der Bevölkerung für die Lebenssituation von Frauen mit genetisch bedingtem Hochrisiko für Brustkrebs geführt. Der folgende Artikel resümiert den heutigen Wissensstand zum Erkrankungsrisiko und zum Vorgehen in der genetischen Beratung, der Testung und der Prophylaxe bei familiär gehäuftem Brustkrebs.
JULIA SCHNABEL, HEIKE HEUER, KONSTANTIN J. DEDES, DANIEL FINK
Nach Bekanntwerden der prophylaktischen Mastektomie bei dem US-amerikanischen Medienstar kam es zu einer öffentlichen Diskussion über die genetische Testung und die prophylaktische Chirurgie. In der klinischen Praxis wurde ein erhöhter Bedarf an medizinischer und genetischer Beratung von erkrankten Patientinnen, von Frauen mit und sogar ohne Familienbelastung wahrgenommen. Dieses Erstgespräch in der gynäkologischen Praxis umfasst I einerseits die Identifikation von Hochrisikofami-
lien zur weiteren Beratung und Testung I andererseits die Aufklärung von Frauen ohne fa-
miliäre Belastung über ihr natürliches Risiko und die empfohlenen Screeninguntersuchungen.
Das hereditäre Mammakarzinom
Das Lebenszeitrisiko einer Frau, an einem Mammakarzinom zu erkranken, beträgt 10 bis 12%. Bei zirka 20% der Mammakarzinomfälle zeigt sich eine familiäre Häufung, wovon 5 bis 10% mit monogen vererbten, hoch penetranten Genen assoziiert sind. Bei der Hälfte davon handelt es sich um Mutationen in den BRCA-1- und BRCA-2-Genen (= BReastCAncer), auch als HBOC-Syndrom (hereditary breast and ovarian cancer syndrome) bezeichnet (1). Weitere Mutationen sind bekannt: RAD51C, auch als BRCA-3 bezeichnet (2), TP53 (Li-Fraumeni-Syndrom), STK11 (Peutz-Jeghers-Syndrom), MLH1/MSH2/MSH6 (hereditary nonpolyposis colorectal cancer [HNPCC]Syndrome) sowie PTEN (Cowden-Syndrom). Weitere
* (Kluger J et al.: «The Angelina effect». Time Magazine, Mai 2013. Artikel nach prophylaktischer Brustektomie bei der amerikanischen Schauspielerin Angelina Jolie.)
Gene werden anhand der Familienstammbäume vermutet, konnten jedoch noch nicht identifiziert werden. Der kombinierten Wirkung von Genen, die mit moderaten und niedrigen Brustkrebsrisiken assoziiert sind, sogenannten polygenen Erbgängen, wird ein Grossteil der familiär bedingten Mammakarzinome zugesprochen (3, 4) (Tabelle 1). In der Routinediagnostik spielen diese Risikogene aktuell noch keine Rolle. BRCA-1- und BRCA-2-Mutationen werden autosomal dominant vererbt, das heisst sowohl über die mütterliche als auch über die väterliche Linie; somit sind beide Familienstammbäume bei der Beratung und der Testung zu berücksichtigen. Das Risiko, den Gendefekt den Nachkommen zu vererben, beträgt 50%. BRCA-1 und BRCA-2 sind Tumorsuppressorgene: Erst wenn das zweite gesunde Wild-Typ-Allel mutiert – die Wahrscheinlichkeit nimmt mit steigendem Alter zu – kommt es zum Krankheitsausbruch. Das Risiko für die assoziierte Entwicklung eines Mamma- oder Ovarialkarzinoms zeigt demnach eine altersabhängige Kumulation. Männer zeigen eine niedrigere genetische Penetranz, daher ergeben sich teilweise auch Schwierigkeiten bei der Interpretation des Stammbaumes.
Klinische und morphologische Charakteristika beim BRCA-mutierten Mammakarzinom
In der klinischen Praxis zeigen sich folgende Charakteristika: I Es besteht eine familiäre Häufung BRCA-assozi-
ierter Tumore (der Mamma, des Ovars, des Pankreas, der Prostata).
12 GYNÄKOLOGIE 3/2014
SCHWERPUNKT
I Das Ersterkrankungsalter von Mutationsträgerinnen liegt rund 20 Jahre vor demjenigen von Frauen mit sporadischem Mammakarzinom.
I Es besteht ein erhöhtes Risiko für ein kontralaterales Mammakarzinom: Das kumulative Erkrankungsrisiko liegt hier bei 47,4% (95%-KI: 38,8%– 56,0%), abhängig vom Ersterkrankungsalter und dem betroffenen BRCA-Gen. BRCA-1-Mutationsträgerinnen haben ein 1,6-fach höheres Risiko als BRCA-2-Mutationsträgerinnen (5).
I Bei Mammakarzinomen bei BRCA-1-Mutationsträgerinnen bestehen häufig ein tripelnegativer Immunphänotyp und eine hohe Proliferation. Die Tumoren metastasieren überwiegend in den ersten drei Jahren nach Diagnosestellung (6, 7).
I Die Tumoren bei BRCA-2-Mutationsträgerinnen verhalten sich morphologisch ähnlich dem sporadischen, hormonrezeptorpositiven Mammakarzinom.
Erkrankungsrisiko
Bei Nachweis einer Mutation eines der Hochpenetranzgene (BRCA-1, BRCA-2, RAD51C ) steigt das Erkrankungsrisiko um ein Vielfaches. Abhängig von der Anzahl betroffener Angehöriger zeigt sich ein weiterer Risikoanstieg (1). Das genaue Risiko, als Mutationsträgerin ein Mamma- oder ein Ovarialkarzinom zu entwickeln, ist nicht bekannt. Selbst bei gleichen Mutationen in Familien gleicher ethnischer Herkunft ist das individuelle Krankheitsrisiko unterschiedlich. Die Schätzwerte stammen einerseits von Familien mit zahlreichen Betroffenen, andererseits von Familien mit nur wenigen Betroffenen oder aus bevölkerungsbasierten Studien. Daher sind in der Literatur häufig sehr unterschiedliche Zahlen zu finden, welche teilweise ein Lebenszeitrisiko für Mammakarzinom über 85% zeigen. Eine Metaanalyse von 2007 zeigt ein kumulatives Erkrankungsrisiko bis zum 70. Lebensjahr für ein Mammakarzinom von 57% bei BRCA-1- und 49% für BRCA-2-Mutationsträgerinnen sowie von 40% respektive von 18% für ein Ovarialkarzinom (Tabelle 2). Grundsätzlich sollte bei der Beratung daher das individuelle Risiko im Vordergrund stehen und nicht das Lebenszeitrisiko.
Tabelle 1:
Häufigkeit hoch penetranter Gene in mit Brustkrebs belasteten Familien (27)
BRCA-1 BRCA-2 RAD51C (BRCA-3) P53 PTEN STK11
HBOC-Syndrom HBOC-Syndrom Hereditäres Mammakarzinom Li-Fraumeni-Syndrom Cowden-Syndrom Peutz-Jeghers-Syndrom
~ 30 % ~ 25 % ~ 5% ≤ 1% ≤ 1% ≤ 1%
Genetische Beratung und Testung
Fachübergreifendes interdisziplinäres Beratungskonzept Einer molekulargenetischen Abklärung muss eine Beratung vorausgehen: Diese muss umfassend und professionell erfolgen und wird von Gynäkologen, Genetikern und Psychologen im interdisziplinären Konzept angeboten. Die Entscheidungsfindung für Erkrankte, Ratsuchende und Angehörige kann sich schwierig gestalten, denn ein Mutationsnachweis kann zu einer erheblichen Belastung führen. Psychologische, ethische und gesellschaftliche Aspekte können eine grosse Bedeutung einnehmen. Die vielseitigen Konsequenzen für Betroffene und ihre Angehörigen müssen vorher eingehend besprochen werden. Eine begleitende psychologische Beratung ist ein wesentlicher Bestandteil.
Indikationen Grundsätzlich soll bei jeder Patientin mit der Diagnose Mamma- oder Ovarialkarzinom die Frage nach einer tumorgenetischen Beratung gestellt werden. Entscheidend für die Beurteilung der Beratungsnotwendigkeit sind das Alter der erkrankten Patientin und die systematische Stammbaumanalyse. Eine genetische Diagnostik wird ab einem Mutationsrisiko von 10% empfohlen. Das empirische Mutationsrisiko kann basierend auf den Daten des Deutschen Konsortiums für familiären Brust- und Eierstockkrebs abgelesen werden (Tabelle 3). Im Rahmen des Beratungsgesprächs sollte zunächst eine detaillierte Stammbaumanalyse erfolgen; hier handelt es sich um das wichtigste Instrument zur Evaluation des Risikos und der Entscheidung für oder gegen die Empfehlung einer genetischer Testung. Diese Beratung sollte in der Schweiz in einem der
Tabelle 2:
Kumulatives Risiko bis zum 70. Lebensjahr bei BRCA-Mutationsträgern (28, 29)
Mammakarzinom (Frau) Ovarialkarzinom Mammakarzinom (Mann)
Risiko der Allgemeinbevölkerung 10–12% 1,4% 0,1%
Bei BRCA-1-Mutationen 57% 40% 1%
Bei BRCA-2-Mutationen 49% 18% 6%
GYNÄKOLOGIE 3/2014
13
SCHWERPUNKT
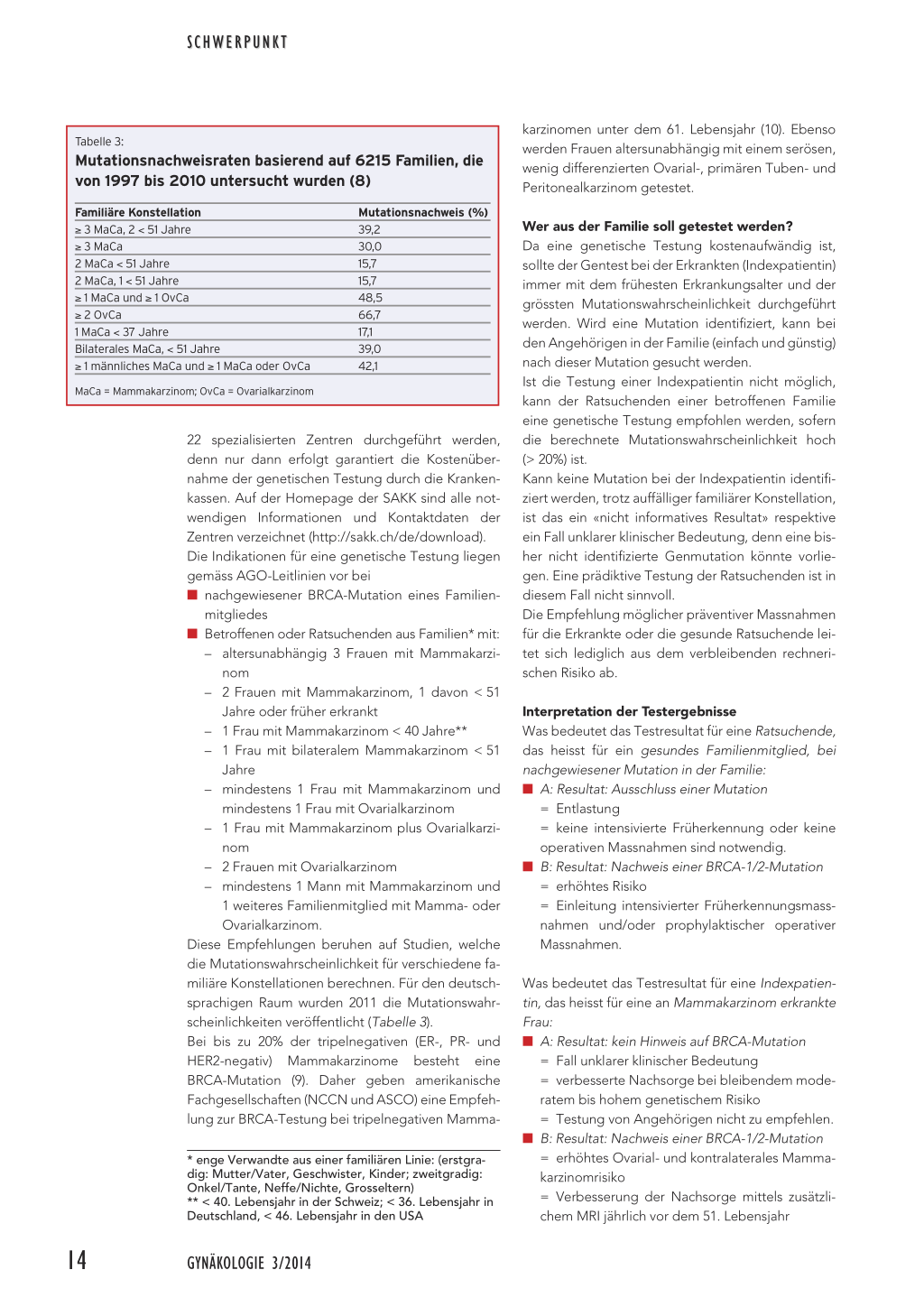
Tabelle 3:
Mutationsnachweisraten basierend auf 6215 Familien, die von 1997 bis 2010 untersucht wurden (8)
Familiäre Konstellation
≥ 3 MaCa, 2 < 51 Jahre ≥ 3 MaCa 2 MaCa < 51 Jahre 2 MaCa, 1 < 51 Jahre ≥ 1 MaCa und ≥ 1 OvCa ≥ 2 OvCa 1 MaCa < 37 Jahre Bilaterales MaCa, < 51 Jahre ≥ 1 männliches MaCa und ≥ 1 MaCa oder OvCa
MaCa = Mammakarzinom; OvCa = Ovarialkarzinom
Mutationsnachweis (%) 39,2 30,0 15,7 15,7 48,5 66,7 17,1 39,0 42,1
22 spezialisierten Zentren durchgeführt werden, denn nur dann erfolgt garantiert die Kostenübernahme der genetischen Testung durch die Krankenkassen. Auf der Homepage der SAKK sind alle notwendigen Informationen und Kontaktdaten der Zentren verzeichnet (http://sakk.ch/de/download). Die Indikationen für eine genetische Testung liegen gemäss AGO-Leitlinien vor bei I nachgewiesener BRCA-Mutation eines Familien-
mitgliedes I Betroffenen oder Ratsuchenden aus Familien* mit:
– altersunabhängig 3 Frauen mit Mammakarzinom
– 2 Frauen mit Mammakarzinom, 1 davon < 51 Jahre oder früher erkrankt
– 1 Frau mit Mammakarzinom < 40 Jahre** – 1 Frau mit bilateralem Mammakarzinom < 51
Jahre – mindestens 1 Frau mit Mammakarzinom und
mindestens 1 Frau mit Ovarialkarzinom – 1 Frau mit Mammakarzinom plus Ovarialkarzi-
nom – 2 Frauen mit Ovarialkarzinom – mindestens 1 Mann mit Mammakarzinom und
1 weiteres Familienmitglied mit Mamma- oder Ovarialkarzinom. Diese Empfehlungen beruhen auf Studien, welche die Mutationswahrscheinlichkeit für verschiedene familiäre Konstellationen berechnen. Für den deutschsprachigen Raum wurden 2011 die Mutationswahrscheinlichkeiten veröffentlicht (Tabelle 3). Bei bis zu 20% der tripelnegativen (ER-, PR- und HER2-negativ) Mammakarzinome besteht eine BRCA-Mutation (9). Daher geben amerikanische Fachgesellschaften (NCCN und ASCO) eine Empfehlung zur BRCA-Testung bei tripelnegativen Mamma-
* enge Verwandte aus einer familiären Linie: (erstgradig: Mutter/Vater, Geschwister, Kinder; zweitgradig: Onkel/Tante, Neffe/Nichte, Grosseltern) ** < 40. Lebensjahr in der Schweiz; < 36. Lebensjahr in Deutschland, < 46. Lebensjahr in den USA
karzinomen unter dem 61. Lebensjahr (10). Ebenso werden Frauen altersunabhängig mit einem serösen, wenig differenzierten Ovarial-, primären Tuben- und Peritonealkarzinom getestet.
Wer aus der Familie soll getestet werden? Da eine genetische Testung kostenaufwändig ist, sollte der Gentest bei der Erkrankten (Indexpatientin) immer mit dem frühesten Erkrankungsalter und der grössten Mutationswahrscheinlichkeit durchgeführt werden. Wird eine Mutation identifiziert, kann bei den Angehörigen in der Familie (einfach und günstig) nach dieser Mutation gesucht werden. Ist die Testung einer Indexpatientin nicht möglich, kann der Ratsuchenden einer betroffenen Familie eine genetische Testung empfohlen werden, sofern die berechnete Mutationswahrscheinlichkeit hoch (> 20%) ist. Kann keine Mutation bei der Indexpatientin identifiziert werden, trotz auffälliger familiärer Konstellation, ist das ein «nicht informatives Resultat» respektive ein Fall unklarer klinischer Bedeutung, denn eine bisher nicht identifizierte Genmutation könnte vorliegen. Eine prädiktive Testung der Ratsuchenden ist in diesem Fall nicht sinnvoll. Die Empfehlung möglicher präventiver Massnahmen für die Erkrankte oder die gesunde Ratsuchende leitet sich lediglich aus dem verbleibenden rechnerischen Risiko ab.
Interpretation der Testergebnisse Was bedeutet das Testresultat für eine Ratsuchende, das heisst für ein gesundes Familienmitglied, bei nachgewiesener Mutation in der Familie: I A: Resultat: Ausschluss einer Mutation
= Entlastung = keine intensivierte Früherkennung oder keine operativen Massnahmen sind notwendig. I B: Resultat: Nachweis einer BRCA-1/2-Mutation = erhöhtes Risiko = Einleitung intensivierter Früherkennungsmassnahmen und/oder prophylaktischer operativer Massnahmen.
Was bedeutet das Testresultat für eine Indexpatientin, das heisst für eine an Mammakarzinom erkrankte Frau: I A: Resultat: kein Hinweis auf BRCA-Mutation
= Fall unklarer klinischer Bedeutung = verbesserte Nachsorge bei bleibendem moderatem bis hohem genetischem Risiko = Testung von Angehörigen nicht zu empfehlen. I B: Resultat: Nachweis einer BRCA-1/2-Mutation = erhöhtes Ovarial- und kontralaterales Mammakarzinomrisiko = Verbesserung der Nachsorge mittels zusätzlichem MRI jährlich vor dem 51. Lebensjahr
14 GYNÄKOLOGIE 3/2014
SCHWERPUNKT
Tabelle 4:
Intensivierte Früherkennungsmassnahmen
Ärztliche Tastuntersuchung alle 6 Monate Mammasonografie alle 6 Monate Mammografie alle 12 Monate Zyklusadaptiertes MRI der Brust alle 12 Monate
* Oder 5 Jahre vor dem frühesten Erkrankungsalter
ab dem 25. Lebensjahr* ab dem 25.*–55. Lebensjahr ab dem 30.–35. Lebensjahr* ab dem 25. Lebensjahr*
= gegebenenfalls prophylaktische Operationen. Bei einem nicht informativen Testresultat ist die mathematische Risikoberechnung für die Trägerwahrscheinlichkeit einer Mutation respektive die altersspezifische Erkrankungswahrscheinlichkeit anhand von Computerprogrammen möglich. Abhängig vom Programm fliessen Informationen wie Anzahl der familiären Tumorerkrankungen, Verwandtschaftsgrad und Erkrankungsalter in die Kalkulation ein.
Präventive Massnahmen
Intensivierte Früherkennung Für Frauen mit einem Mutationsnachweis und einem Erkrankungsrisiko von mehr als 30% oder einem geschätzten Mutationsrisiko (sog. Heterozygotierisiko) von mehr als 20% ohne Gentest wird ein strukturiertes Früherkennungsprogramm empfohlen. Aufgrund des jungen Erkrankungsalters müssen Früherkennungsmassnahmen vor dem Beginn des für die weibliche Allgemeinbevölkerung empfohlenen Mammografiescreenings begonnen werden. Effektive Früherkennungsmassnahmen für ein Ovarialkarzinom existieren nicht (11). Diverse Studien konnten weder in der Allgemeinbevölkerung noch in Risikopopulationen eine Evidenz für ein Screening mit transvaginalem Ultraschall und CA-125-Bestimmung nachweisen. Dennoch werden in den USA gemäss Empfehlung der NCCN und ACOG halbjährlich der transvaginale Ultraschall und die CA125-Bestimmung bei Mutationsträgerinnen ab dem 30. beziehungsweise 35. Lebensjahr durchgeführt (Tabelle 4).
Intensivierte Nachsorge bei erkrankten Mutationsträgerinnen Eine jährliche Magnetresonanztomografie (MRT) der Brust wird bei Frauen vor dem 51. Lebensjahr zusätzlich zur Mammografie empfohlen. Die Überlegenheit der MRT gegenüber der Mammografie bei Frauen mit BRCA-Mutation konnte in Studien belegt werden (12).
Prophylaktische Operationen bei nachgewiesener BRCA-Mutation I Prophylaktische bilaterale Mastektomie (PBM) Mit der bilateralen Mastektomie wird eine Senkung des Risikos für eine Brustkrebserkrankung um über
95% und in der Folge eine Senkung der brustkrebsspezifischen Letalität um 90% erreicht (13, 14). Eine PBM sollte erst ab dem 25. Lebensjahr durchgeführt werden (15). Bei der präoperativen Beratung sollte die hetero- oder die autologe Sofortrekonstruktion unbedingt diskutiert werden.
I Bilaterale Salpingo-Oophorektomie (PBSO) Die bilaterale Salpingo-Oophorektomie reduziert das Ovarialkarzinomrisiko um 97% und sollte ausdrücklich empfohlen werden. Zusätzlich wird das Brustkrebsrisiko um 50% (16) und das Risiko für ein kontralaterales Zweitkarzinom um 30 bis 50 % gesenkt (17). Empfohlener Zeitpunkt ist das 40. Lebensjahr respektive sobald die Familienplanung abgeschlossen ist. Eine Hormonersatztherapie wird dann bis zum 50. Lebensjahr empfohlen.
I Kontralaterale oder bilaterale Mastektomie als Sekundärprophylaxe
Die Datenlage ist unklar (18, 19), da trotz signifikanter Reduktion von ipsi- oder kontralateralen Zweitkarzinomen (5, 20) bisher kein Überlebensvorteil durch die radikalere lokale Therapie bewiesen werden konnte (21). In die Beratung sollte das erhöhte Zweiterkrankungsrisiko im Vergleich zu Patientinnen mit sporadischem Mammakarzinom, aber auch die erkrankungsbedingte Prognose der einzelnen Patientin und der bisher fehlende Nachweis eines Überlebensvorteils einfliessen. Alter bei Auftreten des Erstkarzinoms, betroffenes Gen und die Prognose müssen bei der Beratung und der Entscheidung berücksichtigt werden. Die prophylaktische Mastektomie und die SalpingoOophorektomie sind bei fehlendem Mutationsnachweis in der Regel nicht indiziert. Im Einzelfall kann – in Abhängigkeit vom statistischen individuellen Erkrankungsrisiko und bei Erkrankten bei entsprechender Prognose – eine prophylaktischen Operation in Erwägung gezogen werden. Die Datenlage diesbezüglich ist schlecht (2).
Chemoprävention Aufgrund der limitierten Datenlage kann zum präventiven Einsatz von Tamoxifen, Raloxifen und Aromataseinhibitoren bei Mutationsträgerinnen ausserhalb von klinischen Studien zurzeit keine Empfehlung gegeben werden (22). Einen Benefit scheint es für BRCA-2-Mutationsträgerinnen zu geben (diese zeigen ein ähnliches Profil wie Erkrankte mit sporadischen Mammakarzinomen und häufig eine Hormonrezeptorexpression); jedoch ist auch hier die Datenlage aufgrund kleiner Fallzahlen unsicher (23).
16 GYNÄKOLOGIE 3/2014
SCHWERPUNKT
Therapeutische Konsequenzen bei BRCA-Mutation?
Die Therapieverfahren bei sporadischen oder here-
ditären Mamma- und Ovarialkarzinomen unterschie-
den sich bisher nicht. Retrospektive Studien belegen
eine erhöhte Sensitivität der BRCA-assoziierten Tu-
moren gegenüber Platinderivaten (24), prospektive
Studien wurden daraufhin eingeleitet. Noch besteht
keine ausreichende Evidenz für den routinemässigen
Einsatz.
Als vielversprechend gilt bei BRCA-mutierten Karzi-
nomen die zielgerichtete Therapie mit PARP-Inhibi-
toren (PARPi), deren Wirksamkeit bereits in Phase-II-
Studien belegt werden konnte. PARP sind an
DNA-Reparaturmechanismen beteiligt. PARPi zeig-
ten eine hohe Selektivität für BRCA-mutierte Karzi-
nomzellen. Durch Inhibition des Reparaturmechanis-
mus geht die Tumorzelle zugrunde. Dieser Mecha-
nismus wird als «synthetische Letalität» bezeichnet
(25, 26).
Somit wird zukünftig ein BRCA-Mutationsnachweis
auch eine therapeutische Konsequenz darstellen und
das Spektrum als Indiktion für eine genetische Ab-
klärung erweitern.
I
Dr. med. Julia Schnabel (Korrespondenzadresse) Klinik für Gynäkologie UniversitätsSpital Zürich 8061 Zürich E-Mail: julia.schnabel@usz.ch
Interessenkonflikte in Zusammenhang mit diesem Artikel: keine.
Merkpunkte
I Etwa 5% aller Mammakarzinome sind BRCA-bedingt. I Die Beratung bei familiärer Belastung sollte in ei-
nem der 22 spezialisierten interdisziplinären Zentren erfolgen (siehe: www.sakk.ch). I Im Falle einer BRCA-Mutation beträgt das Lebenszeitrisiko für ein Mammakarzinom bis zu 85% und für ein Ovarialkarzinom bis zu 50%. I Bei nachgewiesener Mutation beziehungsweise Hochrisikokonstellation (Heterozygotenrisiko > 20% oder Lebenszeitrisiko > 30%) ist ein risikoadaptiertes, intensiviertes Früherkennungsprogramm indiziert.
Quellen: 1. Thomssen C, Wand D.: Hereditärer Brustkrebs. in: Onkologe 2012; 18: 216–23. 2. Meindl A, Ditsch N, Kast K, Rhiem K, Schmutzler RK.: Hereditary breast and ovarian cancer – new genes, new treatments, new concepts. Dtsch Arztebl Int 2011; 108(19): 323–30. 3. Ripperger T, Gadzicki D, Meindl A, Schlegelberger B.: Breast cancer susceptibility: current knowledge and implications for genetic counselling. Eur J Hum Genet 2009; 17: 722–31. 4. Turnbull C, Rahman N.: Genetic predisposition to breast cancer: past, present and future. Ann Rev Genomics Hum Genet 2008; 9: 321–45. 5. Graeser MK, Engel Ch, Rhiem K, et al.: Contralateral breast cancer risk in BRCA1 and BRCA2 mutation carriers. J Clin Oncol 2009; 27: 1–6. 6. Lakhani SR, Reis-Filho JS, Fulford L, et al.: Prediction of BRCA1status in patients with breast cancer using estrogen receptor and basal phenotype. Clin Cancer Res 2005; 11: 5175–80. 7. Dent R, Trudeau M, Pritchard KI, et al.: Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13: 4429–34. 8. Schmutzler R.: Breast Cancer Risk and Prevention: Empfehlungen Gynäkologische Onkologie. AGO Kommission Mamma Kapitel 16. (Version 11.1.0, Juli 2011). 9. Gonzalez-Angulo AM, Timms KM, Liu S, et al.: Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res 2011; 17: 1082. 10. NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk assessment: Breast and Ovarian. Version 4.2013. 11. Bosse K.: Screening for ovarian cancer by transvaginal ultrasound and serum CA125 measurement in women with a familial predisposition: a prospective cohort study. Gynecol Oncol 2006; 103(3): 1077–82. 12. Kuhl C.: Prospective multicenter cohort study to refine management recommendations for women at elevated familial risk of breast cancer: the EVA trial. J Clin Oncol 2010; 28(9): 1450–57. 13. Rebbeck TR, Friebel T, Lynch HT.: Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers. The PROSE Study Group. J Clin Oncol 2004; 22: 1055–62. 14. Domchek SM, Friebel TM, Neuhausen SL, et al.: Mortality after bilateral salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers: a prospetive cohort study. Lancet 2006; 7: 223–29. 15. Albrecht U.: Stufe-3-Leitlinie Früherkennung, Diagnostik und Therapie des Mammakarzinoms. 2008. 16. Kauff ND, Domchek SM, Friebel TM, et al.: Risk reducing Salpingo-oophorectomy for the prevention of BRCA1- and BRCA2-associated Breast and Gynecologic Cancer: A multicenter, prospective Study. J Clin Oncol 2008; 26: 1331–37. 17. Metcalfe K, Lynch HT, Ghadirian P.: Contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. J Clin Oncol 2004; 22: 2328–35. 18. Garcia-Etienne CA, Barile M, Gentilini OD et al.: Breast-conserving surgery in BRCA1/2 mutation carriers: are we approaching an answer? Ann Surg Oncol 2009; 16(12): 3380–87. 19. Kirova YM, Savignoni A, Sigal-Zafrani B et al.: Is the breast-conserving treatment with radiotherapy appropriate in BRCA1/2 mutation carriers? Long-term results and review of the literature. Breast Cancer Res Treat 2010; 120(1): 119–26. (Review). 20. Pierce LJ, Levin AM, Rebbeck TR et al.: Ten year multi-institutional results of breast-conserving surgery and radiotherapy in BRCA1/2-associated stage I/II breast cancer. J Clin Oncol 2006; 24(16): 2437–43. 21. Paradiso A, Formenti S.: Hereditary breast cancer: clinical features and risk reduction strategies. Ann Oncol 2011; 22(Suppl 1): i31–i6. 22. Powles TJ, Ashley S, Tidy A et al.: Twenty-year follow-up of the Royal Marsden randomized, double- blinded tamoxifen breast cancer prevention trial. J Natl Cancer Inst 2007; 99(4): 283–90. 23. King MC, Wieand S, Hale K, et al.: Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA 2001; 286: 2251. 24. Byrski T, Gronwald J, Huzarski T, et al.: Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol 2010; 28: 375–79. 25. Farmer H, et al.: Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434(7035): 917–21. 26. Ashworth A.: A synthetic lethal therapeutic approach: poly (ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 2008; 26: 3785–90. 27. Bürki N et al.: Familiärer Brustkrebs – Diagnose, Beratung, Therapie und Langzeitbetreuung. Zürich 2012. 28. Chen S, Parmigiani G.: Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol 2007; 25: 1329. 29. Liede A, Karlan BY, Narod SA.: Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: a review of the literature. J Clin Oncol 2004; 22: 735.
GYNÄKOLOGIE 3/2014
17