
Transkript
BERICHT
Zystische Fibrose
Neue Perspektiven für die Behandlung von Mukoviszidosepatienten
Vor allem dank der Fortschritte in der Antibiotikatherapie ist die Lebenserwartung bei zystischer Fibrose erheblich gestiegen. An einer Pressekonferenz am Kongress der European Respiratory Society in Berlin informierten Professor Matthias Griese und Dr. med. Dominik Hartl über neue therapeutische Ansätze, von denen man sich weitere Verbesserungen erhofft.
RENATE BONIFER
In den Sechzigerjahren des letzten Jahrhunderts wurden Kinder mit zystischer Fibrose in der Regel nur 5 bis 7 Jahre alt, heute hingegen beträgt die durchschnittliche Lebenserwartung bereits 37 Jahre, und man hofft, dass Kinder, die heute mit zystischer Fibrose zur Welt kommen, noch mit wesentlich mehr Lebensjahren rechnen dürfen. Die Gentherapie, auf die man gerade auch bei Mukoviszidose grosse Erwartungen gesetzt hatte, brachte bisher nicht den erwünschten Erfolg. Zwar schaffte es ein rekombinantes DNA-Fragment (Plasmin™), das via Inhalation in die Lungen von CF-Patienten appliziert wurde, bis in Phase-1-Studien, eine Expression des entsprechenden Gens war jedoch nicht nachweisbar, obwohl man gewisse Veränderungen im Chloridtransport der Nasenschleimhaut beobachtete. Diese Versuche sind vorerst gestoppt, und man versucht, die DNA-Nanopartikel für den Gentransfer zu optimieren. Ebenfalls nahe an der Ursache der zystischen Fibrose, das heisst den genetisch bedingten Fehlfunktionen des Chloridtransportermoleküls in Schleimhautzel-
len, wirken Substanzen, die das defekte Transporterprotein reparieren oder in seiner Funktion unterstützen sollen. Professor Matthias Griese von der Universitätskinderklinik München erwähnte in diesem Zusammenhang die Substanzen PTC124, VX-770, Curcumin und VX-809, die sich in unterschiedlichen Entwicklungsphasen befinden. Andere Substanzen zielen auf die Wiederherstellung des zellulären Salztransports (Denufosol, Bronchitol), den Schleim in den Bronchien (NaCl-7%-Inhalation), die chronische Entzündung (Glutathioninhalation) oder die Infektion mit Pseudomonas aeruginosa (neue Darreichungsformen von Antibiotika, Antikörper). Eine Übersicht der aktuellen Forschungslinien und Studienphasen gibt die Tabelle. Eine neues therapeutisches Ziel könnte die Wiederherstellung der Granulozytenfunktion in der Lunge von Patienten mit zystischer Fibrose werden, berichtete Dr. med. Dominik Hartl. Letztlich bestimmen chronische Lungenerkrankungen die Morbidität und Mortalität von Patienten mit zystischer Fibrose. Die Lungen von CF-Patienten sind infolge mangelnder Immunabwehr bakteriell besiedelt (Pseudomonas aeruginosa).
Worin diese mangelnde Immunantwort gegenüber dem Bakterium begründet ist, war jedoch lange Zeit unklar, denn in der Lunge eines CF-Patienten finden sich in der Regel massenhaft neutrophile Granulozyten, normalerweise hocheffiziente «Bakterienkiller». Schon bald wurde klar, dass lokale Faktoren in der Lunge für das Versagen der Neutrophilen verantwortlich sein mussten, denn aus dem Blut gewonnene Neutrophile unterschieden sich zwischen Gesunden und CF-Patienten hinsichtlich ihrer bakterientötenden Kapazität kaum. In der CF-Lunge mit einer chronischen Infekton durch Pseudomonas aeruginosa hingegen sieht die Sache ganz anders aus: Das Bakterium schädigt die Neutrophilen durch Toxine und andere Substanzen, was zu einem vermehrten Absterben der einst so potenten Bakterienkiller führt. Gleichzeitig wird das Wegräumen der Zelltrümmer durch Makrophagen inhibiert. Inhaltsstoffe und Reste der abgestorbenen Neutrophilen bleiben länger im Gewebe liegen, als das normalerweise der Fall wäre. Doch damit nicht genug. Hartl und seine Kollegen fanden heraus, dass das aus den toten Neutrophilen freigesetzte Enzym Elastase einen für die Anti-BakterienAktivierung der noch intakten Neutrophilen entscheidenden Zellrezeptor abschneidet. Die Folgen: Die Neutrophilen werden quasi «entwaffnet» und können ihrer Funktion als Bakterienkiller nicht mehr gerecht werden. Die abgeschnittenen Rezeptorfragmente ihrerseits binden an inflammatorisch aktivierende Rezeptoren der Schleimhaut und heizen die Migration von Neutrophilen in das Gewebe weiter an, die dort wiederum sterben und ihre noch lebenden Mit-
482 ARS MEDICI 12 ■ 2009
BERICHT
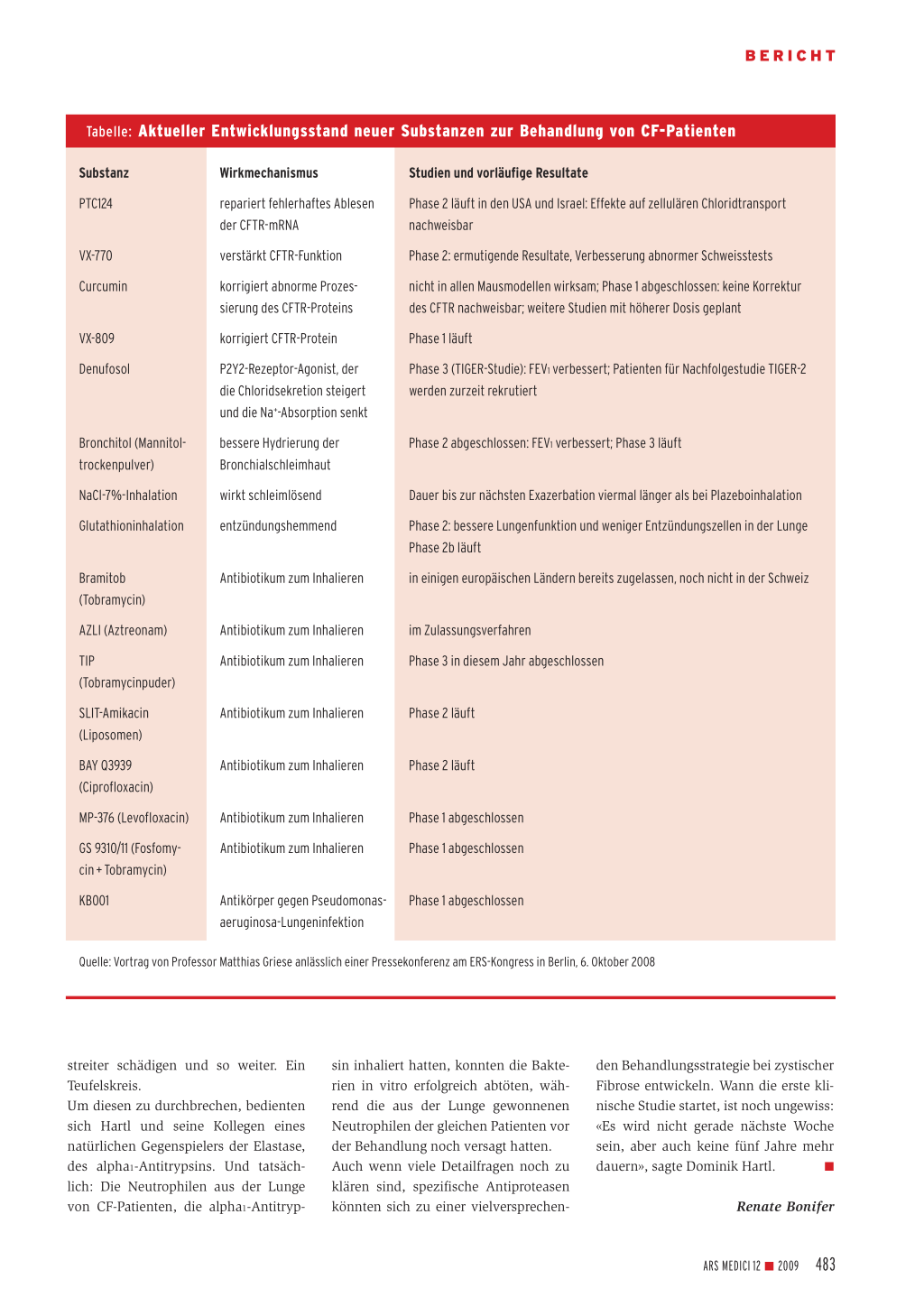
Tabelle: Aktueller Entwicklungsstand neuer Substanzen zur Behandlung von CF-Patienten
Substanz PTC124
VX-770 Curcumin
VX-809 Denufosol
Bronchitol (Mannitoltrockenpulver) NaCl-7%-Inhalation Glutathioninhalation
Bramitob (Tobramycin) AZLI (Aztreonam) TIP (Tobramycinpuder) SLIT-Amikacin (Liposomen) BAY Q3939 (Ciprofloxacin) MP-376 (Levofloxacin) GS 9310/11 (Fosfomycin + Tobramycin) KB001
Wirkmechanismus repariert fehlerhaftes Ablesen der CFTR-mRNA verstärkt CFTR-Funktion korrigiert abnorme Prozessierung des CFTR-Proteins korrigiert CFTR-Protein P2Y2-Rezeptor-Agonist, der die Chloridsekretion steigert und die Na+-Absorption senkt bessere Hydrierung der Bronchialschleimhaut wirkt schleimlösend entzündungshemmend
Antibiotikum zum Inhalieren
Studien und vorläufige Resultate Phase 2 läuft in den USA und Israel: Effekte auf zellulären Chloridtransport nachweisbar Phase 2: ermutigende Resultate, Verbesserung abnormer Schweisstests nicht in allen Mausmodellen wirksam; Phase 1 abgeschlossen: keine Korrektur des CFTR nachweisbar; weitere Studien mit höherer Dosis geplant Phase 1 läuft Phase 3 (TIGER-Studie): FEV1 verbessert; Patienten für Nachfolgestudie TIGER-2 werden zurzeit rekrutiert
Phase 2 abgeschlossen: FEV1 verbessert; Phase 3 läuft
Dauer bis zur nächsten Exazerbation viermal länger als bei Plazeboinhalation Phase 2: bessere Lungenfunktion und weniger Entzündungszellen in der Lunge Phase 2b läuft in einigen europäischen Ländern bereits zugelassen, noch nicht in der Schweiz
Antibiotikum zum Inhalieren Antibiotikum zum Inhalieren
im Zulassungsverfahren Phase 3 in diesem Jahr abgeschlossen
Antibiotikum zum Inhalieren
Phase 2 läuft
Antibiotikum zum Inhalieren
Phase 2 läuft
Antibiotikum zum Inhalieren Antibiotikum zum Inhalieren
Phase 1 abgeschlossen Phase 1 abgeschlossen
Antikörper gegen Pseudomonas- Phase 1 abgeschlossen aeruginosa-Lungeninfektion
Quelle: Vortrag von Professor Matthias Griese anlässlich einer Pressekonferenz am ERS-Kongress in Berlin, 6. Oktober 2008
streiter schädigen und so weiter. Ein Teufelskreis. Um diesen zu durchbrechen, bedienten sich Hartl und seine Kollegen eines natürlichen Gegenspielers der Elastase, des alpha1-Antitrypsins. Und tatsächlich: Die Neutrophilen aus der Lunge von CF-Patienten, die alpha1-Antitryp-
sin inhaliert hatten, konnten die Bakterien in vitro erfolgreich abtöten, während die aus der Lunge gewonnenen Neutrophilen der gleichen Patienten vor der Behandlung noch versagt hatten. Auch wenn viele Detailfragen noch zu klären sind, spezifische Antiproteasen könnten sich zu einer vielversprechen-
den Behandlungsstrategie bei zystischer
Fibrose entwickeln. Wann die erste kli-
nische Studie startet, ist noch ungewiss:
«Es wird nicht gerade nächste Woche
sein, aber auch keine fünf Jahre mehr
dauern», sagte Dominik Hartl.
■
Renate Bonifer
ARS MEDICI 12 ■ 2009 483