




Transkript
ÜBERSICHT
Das blasse Kind
Rationale Anämiediagnostik in der Hausarztpraxis
Die Ursachen der Anämie im Kindesalter, die zur
Vorstellung eines blassen Kindes beim Arzt führen,
sind breit gefächert. Sie reichen von der physio-
logischen Trimenonreduktion über die harmlose
transitorische Erythroblastopenie bis zu genetisch
bedingten lebensbedrohlichen Anämieformen. Das
Spektrum der Anämien bei Kindern unterscheidet sich
in vielen Aspekten erheblich von dem bei Erwachsenen.
Diese Darstellung verschafft einen Überblick über
wichtige Anämien im Kindesalter und bietet ein ratio-
nales diagnostisches Vorgehen zur Annäherung an
die in einigen Bereichen auch diffizile Differenzial-
diagnose auf.
HOLGER N. LODE
Eine Anämie ist über die Erniedrigung der Hämoglobinkonzentration unter den physiologischen Normalwert definiert. Als pathologisch wird im Allgemeinen eine Reduktion um den Betrag von zwei Standardabweichungen (SD) vom Mittelwert einer altersentsprechenden Normalpopulation gewertet. Da die Hämoglobinkonzentration während der Entwicklung eines Kindes von der Neonatalperiode bis ins Erwachsenenalter starken Schwankungen unterliegt, ist die Kenntnis der altersentsprechenden Normwerte eine wesentliche Voraussetzung (Tabelle 1) für die Diagnose einer Anämie (1). Hier ist eine physiologische Abnahme der Hämoglobinkonzentration im dritten bis sechsten Monat hervorzuheben, welche als Trimenonreduktion bezeichnet wird (Tabelle 1). Die Trimenonreduktion hängt ursächlich mit dem Ersatz des fetalen Hämoglobins (HbF) durch das adulte Hämoglobin (HbA) zusammen. Das führt durch die Abnahme der Affinität von Sauerstoff
an das Hämoglobin zu einer verbesserten Sauerstoffabgabe im Gewebe und somit zu einer vermehrten Sauerstoffausschöpfbarkeit, welche sich in einer Verschiebung der Sauerstoffbindungskurve nach rechts widerspiegelt. Dadurch wird die Erythropoetinproduktion nach der Geburt stark gedrosselt und liegt in der Phase der Trimenonreduktion auf minimalem Niveau. Als Folge findet man auch eine erniedrigte Retikulozytenzahl. Die Trimenonreduktion ist also das Ergebnis einer physiologischen funktionellen Regulation, die in aller Regel keiner Therapie bedarf, und nicht Ausdruck einer Insuffizienz. Kenntnisse über diese Zusammenhänge spielen eine wichtige Rolle für eine Einschätzung der Notwendigkeit weiterführender Diagnostik bei einem blassen Säugling.
Symptome Blässe ist das Leitsymptom einer manifesten Anämie, die in aller Regel unter einem Hämoglobinwert von 9 g/dl auftritt. Diese ist bei Kindern mediterraner oder nicht kaukasischer Abstammung auch durch blasse Haut im Bereich der Schleimhäute, insbesondere der Konjunktiven, gut erkennbar. Weitere Zeichen sind Müdigkeit, Abgeschlagenheit, häufig vergesellschaftet mit Konzentrationsschwäche, und Kopfschmerzen. Häufig fallen eine Tachykardie sowie ein systolisches Herzgeräusch über der Aorten- und Pulmonalklappe auf (Nonnensausen, Anämiegeräusch). Die Schwere der Symptomatik hängt im Besonderen von der Geschwindigkeit des Abfalls der Hämoglobinkonzentration ab.
Merksätze
■ Eine rationale Anämiediagnostik kann mit wenigen Schritten kostengünstig zur richtigen Diagnose führen.
■ Nach Anamnese und klinischer Untersuchung können die Beurteilung des maschinellen Blutbilds und die nachfolgende Mikroskopie des Blutausstrichs jede Anämie so weit klassifizieren, dass nur gezielte Untersuchungen zum Beweis der Verdachtsdiagnose notwendig werden.
668 ARS MEDICI 15 ■ 2008
DAS BLASSE KIND
Tabelle 1: Altersabhängige Mittelwerte und Normgrenzen Tabelle 1: der Hämoglobinkonzentration
Alter Hämoglobinkonzentration (g/dl) Verhältnis
Mittelwert (MW)
MW-2XSD* HbF:HbA (%)
Neugeborene 3–6 Monate** 1/2 –6 Jahre 6–12 Jahre Männer Frauen
* Standardabweichung ** Trimenonreduktion
18,5 11,5 12,5 13,5 15,5 14,0
14,5 70:30 9,5 5:95 11,0 1:99 11,5 1:99 13,5 1:99 12,0 1:99
«pure red cell aplasia» Transitorische Erythroblastopenie des Kindesalters
Blackfan-Diamond-Anämie Parvovirus-B19-Infektion Bildungsstörung aller drei Zellreihen
Fanconi-Anämie Aplastische Anämie Störung des Erythropoetins (Epo) Niereninsuffizienz Anämie bei chronischer Erkrankung Hypothyreoidismus, Hypopituitarismus Antikörper gegen Epo und Eporezeptor
Akuter Blutverlust Sequestration
Gefässmalformation Blutverdünnung
Erythrozytenverlust
Verteilungsstörung
angeboren Membrandefekte Enzymdefekte Sichelzellanämie Instabile Hämoglobine
Verkürzte Überlebenszeit
von Erythrozyten
Thalassämie
erworben
Autoimmunhämolytische Anämie
Thermische, toxische Noxen
Mikroangiopathische Formen
(HUS, TTP)
Oxidative Hämoglobinschädigung
Störung erythropoetischer
Progenitoren
Anämie
Verdrängung des
Knochenmarks
angeboren Osteopetrose
erworben Myelofibrose Leukämien Neuroblastom
Abbildung 1: Gliederung der Anämien im Kindesalter nach Ursache
Störung der Hämoglobinproduktion
angeboren Thalassämie Sideroblastische Anämie Instabile Hämoglobine erworben Eisenmangel Bleivergiftung
Störung des DNA-Stoffwechsels
Vitamin B12 Folsäure Orotazidurie
Eisenmangelanämie oder einer transitorischen Erythroblastopenie des Kindesalters bei derselben Hämoglobinkonzentration klinisch kaum beeinträchtigt ist. Bei langsamer Anämisierung stehen dem Organismus eine Reihe von Kompensationsmechanismen zur Verfügung, welche das Auftreten einer klinischen Symptomatik auch bei sehr niedrigen Hämoglobinwerten verringern. Hierzu zählen eine erythropoetinabhängige Neubildung von Erythrozyten sowie eine Optimierung der Sauerstoffabgabe aus dem Hämoglobin in das Gewebe. Dieses wird durch Faktoren erreicht, welche zu einer Verschiebung der Sauerstoffdissoziationskurve nach rechts führen. Hierzu zählt unter anderem die Erhöhung des 2,3-Diphosphoglycerat-Gehalts der Erythrozyten. Ferner tragen eine Steigerung der Atemfrequenz sowie des Herzminutenvolumens zur Kompensation bei. Allerdings sind Hämoglobinwerte unter 5 g/dl auch bei einem gut adaptierten Kind nicht ungefährlich, da es bei plötzlich auftretenden Kreislaufbelastungen, wie zum Beispiel bei einem hoch fieberhaften Infekt, zu einer raschen Dekompensation mit Herz-Kreislauf-Versagen kommen kann.
Ursachen Prinzipiell lassen sich die Ursachen einer Anämie in verminderte Bildung von Hämoglobin oder Erythrozyten und vermehrten Untergang oder Verlust von Erythrozyten einteilen (Abbildung 1). Da Erkrankungen, die eine ineffektive Erythropoese bedingen, oft auch eine verkürzte Lebenszeit von zirkulierenden Erythrozyten verursachen, ist diese Gliederung arbiträr. Das ätiologische Spektrum der Anämie bei Kindern unterscheidet sich in grossem Ausmass von dem bei Erwachsenen. Die Bildungsstörung, erworben oder angeboren, überwiegt gegenüber erhöhtem Verbrauch oder Verlust von Erythrozyten.
Bei einem Kind mit einer hämolytischen Krise bei Sichelzellanämie oder einer akuten Blutung mit raschem Abfall des Hämoglobins können in kurzer Zeit die klinischen Zeichen einer Herzinsuffizienz (Vorwärts- und Rückwärtsversagen) auftreten, während ein Kind mit einer sich langsam entwickelnden
Diagnostik Der laborchemischen Diagnostik einer Anämie geht, wie bei allen andern Erkrankungen auch, die Erhebung der Anamnese und des körperlichen Untersuchungsbefundes voraus. Hierbei geht es insbesondere darum, eine Systemerkrankung
ARS MEDICI 15 ■ 2008 669
ÜBERSICHT
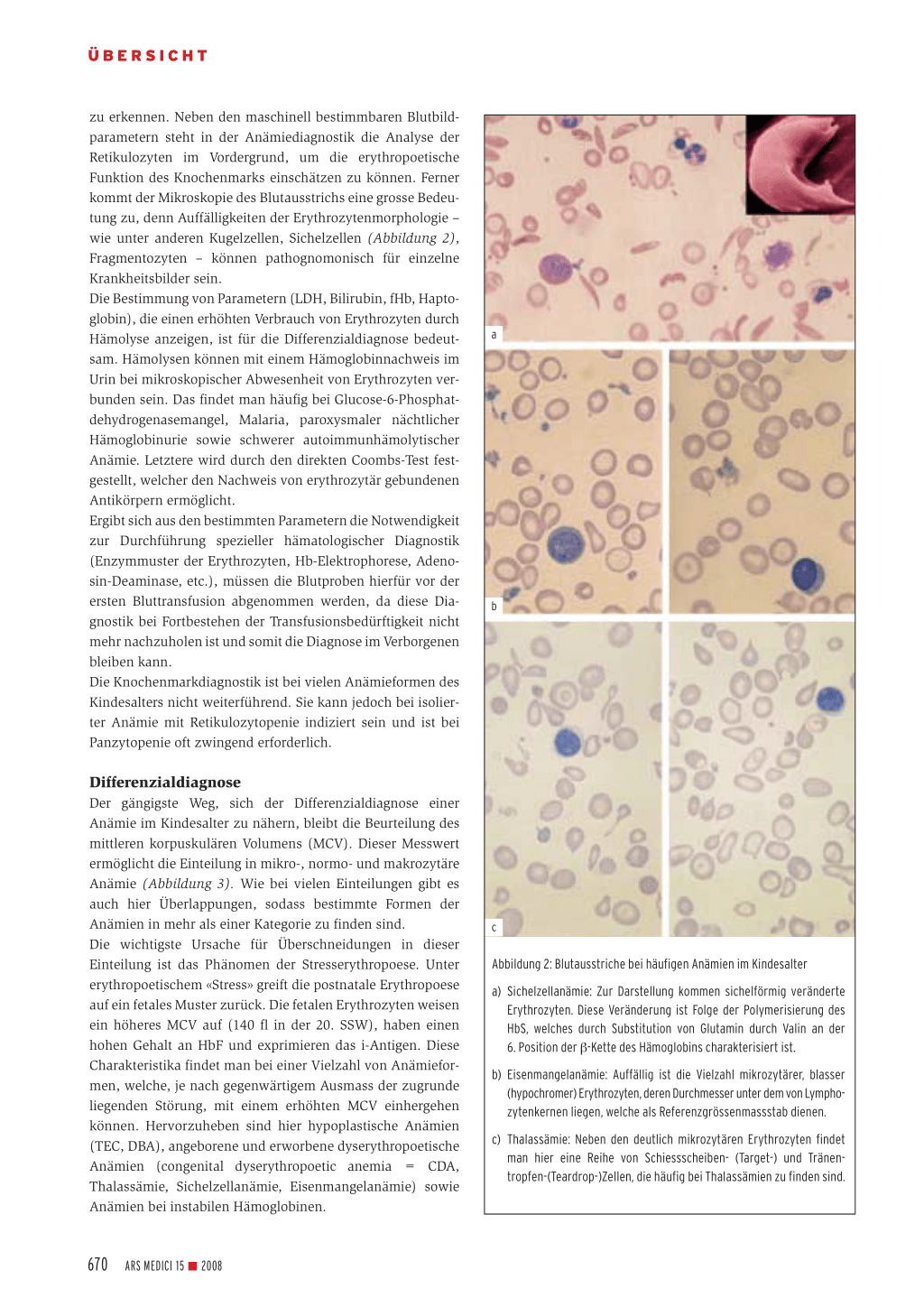
zu erkennen. Neben den maschinell bestimmbaren Blutbildparametern steht in der Anämiediagnostik die Analyse der Retikulozyten im Vordergrund, um die erythropoetische Funktion des Knochenmarks einschätzen zu können. Ferner kommt der Mikroskopie des Blutausstrichs eine grosse Bedeutung zu, denn Auffälligkeiten der Erythrozytenmorphologie – wie unter anderen Kugelzellen, Sichelzellen (Abbildung 2), Fragmentozyten – können pathognomonisch für einzelne Krankheitsbilder sein. Die Bestimmung von Parametern (LDH, Bilirubin, fHb, Haptoglobin), die einen erhöhten Verbrauch von Erythrozyten durch Hämolyse anzeigen, ist für die Differenzialdiagnose bedeutsam. Hämolysen können mit einem Hämoglobinnachweis im Urin bei mikroskopischer Abwesenheit von Erythrozyten verbunden sein. Das findet man häufig bei Glucose-6-Phosphatdehydrogenasemangel, Malaria, paroxysmaler nächtlicher Hämoglobinurie sowie schwerer autoimmunhämolytischer Anämie. Letztere wird durch den direkten Coombs-Test festgestellt, welcher den Nachweis von erythrozytär gebundenen Antikörpern ermöglicht. Ergibt sich aus den bestimmten Parametern die Notwendigkeit zur Durchführung spezieller hämatologischer Diagnostik (Enzymmuster der Erythrozyten, Hb-Elektrophorese, Adenosin-Deaminase, etc.), müssen die Blutproben hierfür vor der ersten Bluttransfusion abgenommen werden, da diese Diagnostik bei Fortbestehen der Transfusionsbedürftigkeit nicht mehr nachzuholen ist und somit die Diagnose im Verborgenen bleiben kann. Die Knochenmarkdiagnostik ist bei vielen Anämieformen des Kindesalters nicht weiterführend. Sie kann jedoch bei isolierter Anämie mit Retikulozytopenie indiziert sein und ist bei Panzytopenie oft zwingend erforderlich.
Differenzialdiagnose Der gängigste Weg, sich der Differenzialdiagnose einer Anämie im Kindesalter zu nähern, bleibt die Beurteilung des mittleren korpuskulären Volumens (MCV). Dieser Messwert ermöglicht die Einteilung in mikro-, normo- und makrozytäre Anämie (Abbildung 3). Wie bei vielen Einteilungen gibt es auch hier Überlappungen, sodass bestimmte Formen der Anämien in mehr als einer Kategorie zu finden sind. Die wichtigste Ursache für Überschneidungen in dieser Einteilung ist das Phänomen der Stresserythropoese. Unter erythropoetischem «Stress» greift die postnatale Erythropoese auf ein fetales Muster zurück. Die fetalen Erythrozyten weisen ein höheres MCV auf (140 fl in der 20. SSW), haben einen hohen Gehalt an HbF und exprimieren das i-Antigen. Diese Charakteristika findet man bei einer Vielzahl von Anämieformen, welche, je nach gegenwärtigem Ausmass der zugrunde liegenden Störung, mit einem erhöhten MCV einhergehen können. Hervorzuheben sind hier hypoplastische Anämien (TEC, DBA), angeborene und erworbene dyserythropoetische Anämien (congenital dyserythropoetic anemia = CDA, Thalassämie, Sichelzellanämie, Eisenmangelanämie) sowie Anämien bei instabilen Hämoglobinen.
a
b
c Abbildung 2: Blutausstriche bei häufigen Anämien im Kindesalter a) Sichelzellanämie: Zur Darstellung kommen sichelförmig veränderte
Erythrozyten. Diese Veränderung ist Folge der Polymerisierung des HbS, welches durch Substitution von Glutamin durch Valin an der 6. Position der β-Kette des Hämoglobins charakterisiert ist. b) Eisenmangelanämie: Auffällig ist die Vielzahl mikrozytärer, blasser (hypochromer) Erythrozyten, deren Durchmesser unter dem von Lymphozytenkernen liegen, welche als Referenzgrössenmassstab dienen. c) Thalassämie: Neben den deutlich mikrozytären Erythrozyten findet man hier eine Reihe von Schiessscheiben- (Target-) und Tränentropfen-(Teardrop-)Zellen, die häufig bei Thalassämien zu finden sind.
670 ARS MEDICI 15 ■ 2008
DAS BLASSE KIND
Makrozytäre Anämien
prägten Hb-Abfällen (Hb < 5 g/dI), die auch über einen länge- Die häufigsten Ursachen einer makrozytären Anämie im Kin- ren Zeitraum hinaus (> 4 Wochen) klinisch symptomatisch
desalter sind Anämien, die mit einer Stresserythropoese ein- sind, sollte mit einer Bluttransfusion behandelt werden. Eine
hergehen (TEC, DBA), sowie der Vitamin-B12- oder Folsäuremangel beziehungsweise diesbezügliche Stoffwechseldefekte.
Indikation zum Einsatz von Steroiden oder Immunsuppressiva gibt es nicht. Die Prognose ist gut, da es sich um eine nach ein
bis zwei Monaten selbstlimitierende Erkrankung durch Bildung
Transitorische Erythroblastopenie des Kindesalters (TEC)
antiidiotypischer Antikörper handelt.
Die TEC ist eine erworbene hypoplastische Anämie, die initial
mit Retikulozytopenie, in der Phase der Regeneration und Diamond-Blackfan-Anämie (DBA)
Stresserythropoese aber mit Retikulozytose einhergeht. In die- Die DBA ist eine angeborene hypoplastische Anämie. Die Dia-
ser Regenerationsphase ist die TEC makrozytär. TEC manifes- gnosestellung erfolgt viel früher als bei der TEC mit einem me-
tiert sich bei Kindern nach einem fieberhaften Infekt mit Blässe. dianen Diagnosealter von 3 Monaten (8). Mehr als 10 Prozent
Sie wurde als häufigste Ursache bei Kindern gefunden, die der Kinder fallen schon bei Geburt auf, und über nahezu 95 Pro-
wegen einer Anämie eine Knochenmarkspunktion erhalten zent der Patienten werden innerhalb der ersten 24 Lebensmo-
hatten (2). Das mediane Erkrankungsalter liegt bei 23 Monaten, nate diagnostiziert. Hämatologisch imponiert eine makrozytäre
auch Säuglinge jünger als 6 Monate sind beschrieben. Da die Anämie. Eine Erhöhung der Adenosin-Deaminase (ADA) in
Hb-Konzentration langsam über Wochen abgefallen ist, sind die Erythrozyten von DBA-Patienten (9) ermöglicht eine Unter-
Kinder meist gut an ihre Anämie adaptiert. Die publizierten scheidung zur TEC.
Inzidenzen von bis zu 5 pro 1 000 000 Kinder (3, 4) unterschät- Im Vordergrund stehen bei Diagnose die Symptome der Anämie
zen die Häufigkeit der Erkrankung vermutlich, da weniger aus- wie Blässe, Müdigkeit und Lustlosigkeit sowie Tachykardie bis
geprägte Anämien übersehen werden. 25 Prozent der Betroffe- hin zum Herzversagen. Fehlbildungen finden sich bei etwa
nen werden erst in der Erholungsphase diagnostiziert, in der 25 bis 40 Prozent der DBA-Patienten (Tabelle 2). Ein Grossteil
bereits eine Retikulozytose (5) sowie Parameter der Stress- dieser Auffälligkeiten betrifft den Kopf und/oder die obere
erythropoese festzustellen sind. Bei wenigen TEC-Patienten Extremität. Ein typisches DBA-Gesicht mit Stupsnase, Hyper-
wurden auch vorübergehende neurologische Symptome wie telorismus, kräftiger Oberlippe und einem intelligenten
Krampfanfälle oder Hemiparesen beschrieben (6, 7). Diese Gesichtsausdruck wurde von Cathie (10) beschrieben (Abbil-
nur wenige Stunden oder Tage anhaltenden Symptome sind dung 4). Daneben können weitere faziale und kraniale An-
möglicherweise ähnlich wie die gelegentlich begleitende Neu- omalitäten bestehen (Mikrognathie, Mikro- oder Makrozepha-
tro- und Thrombozytopenie auf einen gemeinsamen Pathome- lie, Makroglossie, grosse Fontanelle und andere dysmorphe Ver-
chanismus zurückzuführen. Die Ätiologie ist durch postvirale änderungen). Bei etwa 8 Prozent der Patienten finden sich
Prozesse mit Bildung von Antikörpern gegen erythrozytäre Vor- Auffälligkeiten der oberen Extremität, insbesondere triphalan-
läufer charakterisiert, Das Parvo-B19-Virus spielt hier keine geale Daumen (Aase-Syndrom) sowie andere Fehlbildungen
Rolle. Die TEC bedarf in der Regel keiner Therapie. Bei ausge- des radialen Strahls. Vereinzelt sind auch nephrologische und
kardiale Fehlbildungen bei DBA-Patienten be-
schrieben worden. Darüber hinaus wurde ein
gehäuftes Auftreten maligner Erkrankungen bei
Anämie
Patienten mit DBA festgestellt (11). Kleinwuchs bereits bei Geburt fällt bei zirka 10
Prozent der DBA-Patienten auf. Der ausgeprägte
Kleinwuchs stellt für die betroffenen Kinder mit
DBA einen enormen Leidensdruck dar. Durch re-
gelmässige Transfusionen und/oder Steroidthera-
pie erhöht sich der Anteil der kleinwüchsigen
mikrozytär MCV < 70 fl normozytär MCV 70—95 fl makrozytär MCV > 95 fl
DBA-Patienten im Erwachsenenalter auf etwa 50 Prozent. Ungefähr 3 Prozent der DBA-
Patienten sind mental retardiert.
Die DBA folgt bei 30 Prozent der Patienten einem
dominanten Vererbungsmuster, der überwie-
Eisenmangel Thalassämie Anämie chronischer Erkrankungen Sideroblastische Anämie Bleivergiftung
Retikulozytopenie: TEC
Blutverlust Infektanämie Anämie bei chronischer Erkrankung Niereninsuffizienz
Retikulozytose: Sichelzellanämie
Sphärozytose Hämolytisch urämisches
Syndrom Instabile Hämoglobine
Stresserythropoese: Erholung nach TEC oder Chemotherapie oder Blutverlust Diamond-Blackfan-Anämie
Fanconi-Anämie Dyskeratosis congenita
Nutritiver Mangel oder Stoffwechseldefekt von
Vitamin B12 Folsäure
gende Anteil ist also sporadisch (8, 12). Zwei Kandidatengene werden mit dem Auftreten der DBA assoziiert. Mutationen im ribosomalen Protein S19 (RPS 19), welches auf 19q13.2. liegt, führen zu einer reduzierten Proteinsynthese in
spezifischen Zielzellen (13, 14). Der genaue Me-
Abbildung 3: Differenzialdiagnostische Einteilung der Anämie im Kindesalter
chanismus ist allerdings noch unklar. Ein weite-
ARS MEDICI 15 ■ 2008 671
ÜBERSICHT
Tabelle 2: Fehlbildungen bei Tabelle 2: Diamond-Blackfan-Anämie
Region der Fehlbildung
Kopf ohne Augenfehler Augen Hals Daumen Nieren Herz Knochen Andere Mindestens eine Fehlbildung
Häufigkeit
21% 12% 4% 9% 7% 7% 9% 7% 40%
mit charakteristischen Formveränderungen einher, weisen aber dyserythropoetische Veränderungen auf. Die Diagnose wird hier durch weitere Spezialuntersuchungen gestellt. Findet man bei normozytärer Anämie eine Retikulozytopenie, müssen Thrombozyten- und Leukozytenwerte berücksichtigt werden. Sind diese normal, liegt eine TEC ohne Stresserythropoese vor. Differenzialdiagnostisch kommen Anämien bei chronischen Erkrankungen, die nur selten mikrozytär sein können, Infektanämien sowie Anämien bei Niereninsuffizienz infrage. Die beiden Letzteren können auch mit Leuko- oder Thrombozytopenie einhergehen. Liegt eine normozytäre Anämie mit Panzytopenie vor, ändert sich das differenzialdiagnostische Spektrum, und es müssen maligne hämatopoetische Erkrankungen sowie eine Infiltration durch knochenmarkfremde maligne Zellen ausgeschlossen werden.
res DBA-Gen wird auf 8p23.2–p22 vermutet (15). Da es kein ribosomales Protein ist, handelt es sich also um einen zweiten unabhängigen Pathomechanismus. In der Therapie der DBAPatienten werden Kortikosteroide sowie Polytransfusionen eingesetzt. In ausgeprägten Fällen mit Steroidresistenz und hoher Transfusionsfrequenz ist bei Verfügbarkeit eines geeigneten Spenders eine Blutstammzelltransplantation indiziert.
Mikrozytäre Anämien Einer Mikrozytose liegt in der Regel ein Substratmangel für die Hämkomponente oder die Globinketten des Hämoglobins zugrunde. Daher ist nicht nur das MCV erniedrigt, die Erythrozyten sind auch hypochrom. Die weitaus häufigsten Ursachen einer mikrozytären Anämie sind der nutritive Eisenmangel und die Thalassämie.
Vitamin-B12- oder Folsäuremangel Nutritive Ursachen sowie entsprechende Stoffwechseldefekte, die mit einem Mangel an Vitamin B12 oder Folsäure einhergehen, sind die bekanntesten Ursachen für eine makrozytäre Anämie. Die Diagnose wird im Blutausstrich über den Nachweis hypersegmentierter Granulozyten gesichert (16). Megaloblastäre Veränderungen, also Störungen der Kernreifung mit morphologisch aufgetriebenen Zellkernen, findet man im Knochenmark sowohl in erythropoetischen als auch in myeloischen Vorläuferzellen.
a
b
Normozytäre Anämien Die Differenzialdiagnose der normozytären Anämie wird durch die Berücksichtigung der Retikulozytenzahl erleichtert (Abbildung 3). Einer normozytären Anämie mit Retikulozytose liegt meist eine Hämolyse zugrunde. Die mikroskopische Beurteilung des Blutausstrichs ist an dieser Stelle wesentlich, um typische morphologische Veränderungen der Erythrozyten zu erfassen. Beispielhaft hierfür sind die Sichelzellen gezeigt (Abbildung 2a). Der Nachweis von Sphärozyten und Elliptozyten ist genauso richtungsweisend, wobei die Sphärozyten nicht nur bei der hereditären Sphärozytose nachweisbar sind, sondern auch bei der autoimmunhämolytischen Anämie oder beim Morbus Wilson vorkommen können. Anämien bei Enzymdefekten oder instabilen Hämoglobinen gehen nicht
c
Abbildung 4: Diamond-Blackfan-Anämie a) Im Blutausstrich aufgrund der Stresserythropoese überwiegen makrozytäre Erythro-
zyten, die einen grösseren Durchmesser aufweisen als Zellkerne von Lymphozyten (Referenzgrösse). b) «Cathie-Gesicht» (Stupsnase, Hypertelorismus, kräftige Oberlippe, intelligenter Gesichtsausdruck) c) Triphalangeale Daumen (Aase-Syndrom)
672 ARS MEDICI 15 ■ 2008
DAS BLASSE KIND
tion der Globinketten. Im klinischen Alltag ist die
Differenzierung eines Eisenmangels von einer hete-
rozygoten Beta-Thalassämie besonders relevant. Im
Blutausstrich findet man bei der heterozygoten Beta-
Thalassämie vermehrt Schiessscheibenzellen (Ab-
bildung 2c). Hilfreich in der Unterscheidung ist
auch der Thalassämieindex nach Menzer (18, 19).
Dieser bestimmt das Verhältnis von MVC (in fl) zur
Erythrozytenzahl (in 106/µl). Eine Zahl < 13 spricht eher für eine Thalassämie, ein Index >13
für einen Eisenmangel oder andere Hämoglobino-
pathien.
Die Thalassämien sind eine heterogene Gruppe
ab
genetischer Erkrankungen, die aufgrund einer verminderten Syntheserate der Alpha- und Beta-
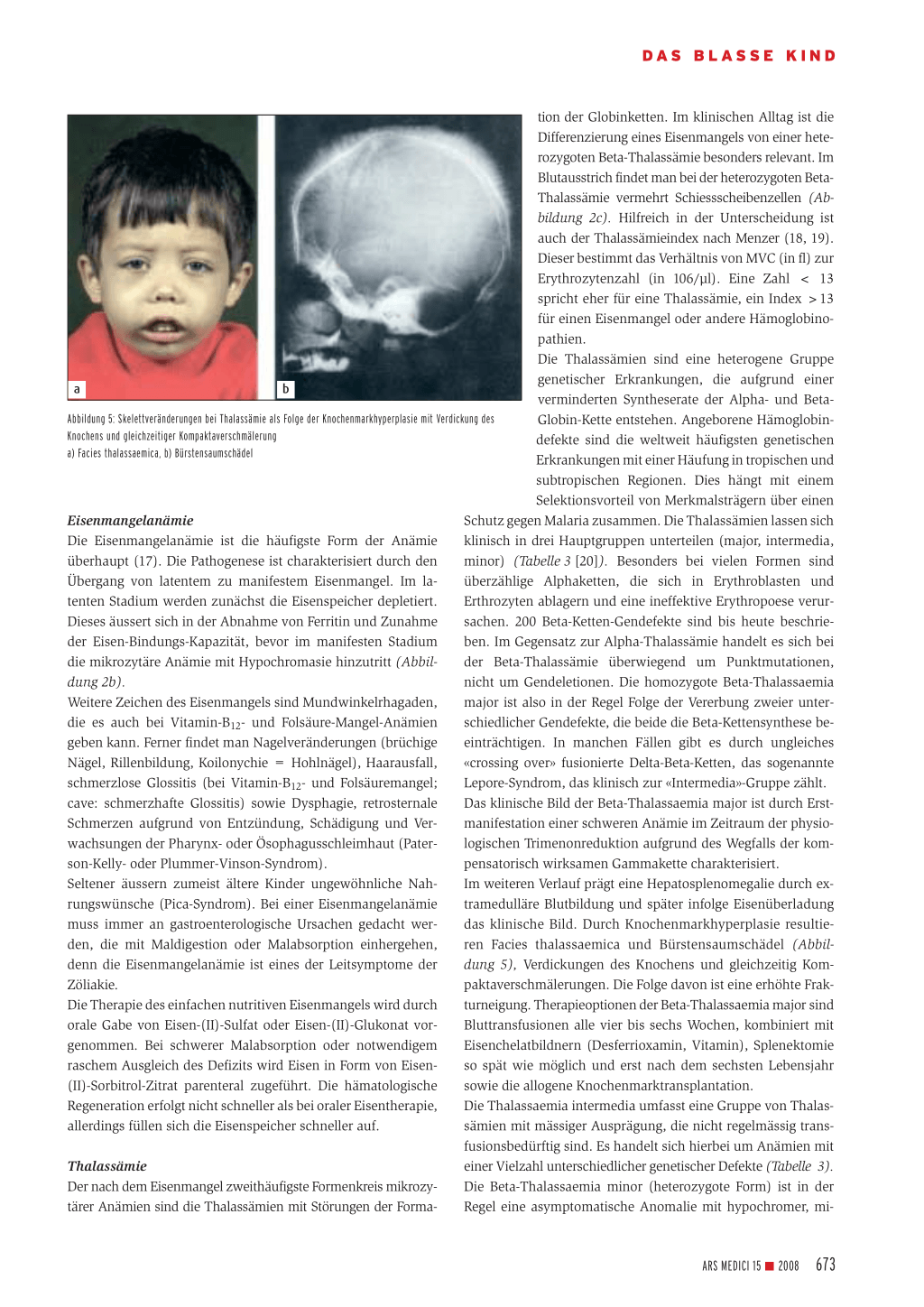
Abbildung 5: Skelettveränderungen bei Thalassämie als Folge der Knochenmarkhyperplasie mit Verdickung des Knochens und gleichzeitiger Kompaktaverschmälerung a) Facies thalassaemica, b) Bürstensaumschädel
Globin-Kette entstehen. Angeborene Hämoglobindefekte sind die weltweit häufigsten genetischen Erkrankungen mit einer Häufung in tropischen und
subtropischen Regionen. Dies hängt mit einem
Selektionsvorteil von Merkmalsträgern über einen
Eisenmangelanämie
Schutz gegen Malaria zusammen. Die Thalassämien lassen sich
Die Eisenmangelanämie ist die häufigste Form der Anämie klinisch in drei Hauptgruppen unterteilen (major, intermedia,
überhaupt (17). Die Pathogenese ist charakterisiert durch den minor) (Tabelle 3 [20]). Besonders bei vielen Formen sind
Übergang von latentem zu manifestem Eisenmangel. Im la- überzählige Alphaketten, die sich in Erythroblasten und
tenten Stadium werden zunächst die Eisenspeicher depletiert. Erthrozyten ablagern und eine ineffektive Erythropoese verur-
Dieses äussert sich in der Abnahme von Ferritin und Zunahme sachen. 200 Beta-Ketten-Gendefekte sind bis heute beschrie-
der Eisen-Bindungs-Kapazität, bevor im manifesten Stadium ben. Im Gegensatz zur Alpha-Thalassämie handelt es sich bei
die mikrozytäre Anämie mit Hypochromasie hinzutritt (Abbil- der Beta-Thalassämie überwiegend um Punktmutationen,
dung 2b).
nicht um Gendeletionen. Die homozygote Beta-Thalassaemia
Weitere Zeichen des Eisenmangels sind Mundwinkelrhagaden, major ist also in der Regel Folge der Vererbung zweier unter-
die es auch bei Vitamin-B12- und Folsäure-Mangel-Anämien geben kann. Ferner findet man Nagelveränderungen (brüchige
schiedlicher Gendefekte, die beide die Beta-Kettensynthese beeinträchtigen. In manchen Fällen gibt es durch ungleiches
Nägel, Rillenbildung, Koilonychie = Hohlnägel), Haarausfall, «crossing over» fusionierte Delta-Beta-Ketten, das sogenannte
schmerzlose Glossitis (bei Vitamin-B12- und Folsäuremangel; cave: schmerzhafte Glossitis) sowie Dysphagie, retrosternale
Lepore-Syndrom, das klinisch zur «Intermedia»-Gruppe zählt. Das klinische Bild der Beta-Thalassaemia major ist durch Erst-
Schmerzen aufgrund von Entzündung, Schädigung und Ver- manifestation einer schweren Anämie im Zeitraum der physio-
wachsungen der Pharynx- oder Ösophagusschleimhaut (Pater- logischen Trimenonreduktion aufgrund des Wegfalls der kom-
son-Kelly- oder Plummer-Vinson-Syndrom).
pensatorisch wirksamen Gammakette charakterisiert.
Seltener äussern zumeist ältere Kinder ungewöhnliche Nah- Im weiteren Verlauf prägt eine Hepatosplenomegalie durch ex-
rungswünsche (Pica-Syndrom). Bei einer Eisenmangelanämie tramedulläre Blutbildung und später infolge Eisenüberladung
muss immer an gastroenterologische Ursachen gedacht wer- das klinische Bild. Durch Knochenmarkhyperplasie resultie-
den, die mit Maldigestion oder Malabsorption einhergehen, ren Facies thalassaemica und Bürstensaumschädel (Abbil-
denn die Eisenmangelanämie ist eines der Leitsymptome der dung 5), Verdickungen des Knochens und gleichzeitig Kom-
Zöliakie.
paktaverschmälerungen. Die Folge davon ist eine erhöhte Frak-
Die Therapie des einfachen nutritiven Eisenmangels wird durch turneigung. Therapieoptionen der Beta-Thalassaemia major sind
orale Gabe von Eisen-(II)-Sulfat oder Eisen-(II)-Glukonat vor- Bluttransfusionen alle vier bis sechs Wochen, kombiniert mit
genommen. Bei schwerer Malabsorption oder notwendigem Eisenchelatbildnern (Desferrioxamin, Vitamin), Splenektomie
raschem Ausgleich des Defizits wird Eisen in Form von Eisen- so spät wie möglich und erst nach dem sechsten Lebensjahr
(II)-Sorbitrol-Zitrat parenteral zugeführt. Die hämatologische sowie die allogene Knochenmarktransplantation.
Regeneration erfolgt nicht schneller als bei oraler Eisentherapie, Die Thalassaemia intermedia umfasst eine Gruppe von Thalas-
allerdings füllen sich die Eisenspeicher schneller auf.
sämien mit mässiger Ausprägung, die nicht regelmässig trans-
fusionsbedürftig sind. Es handelt sich hierbei um Anämien mit
Thalassämie
einer Vielzahl unterschiedlicher genetischer Defekte (Tabelle 3).
Der nach dem Eisenmangel zweithäufigste Formenkreis mikrozy- Die Beta-Thalassaemia minor (heterozygote Form) ist in der
tärer Anämien sind die Thalassämien mit Störungen der Forma- Regel eine asymptomatische Anomalie mit hypochromer, mi-
ARS MEDICI 15 ■ 2008 673
ÜBERSICHT
Tabelle 3: Klinische Einteilung der Thalassämien
Transfusionsbedürftigkeit
Hydrops fetalis major
Thalassaemia intermedia
minor
α-Thalassämie mit Deletionen von 4 Genen letal
Transfusionsbedürftige
homozygote β0-Thalassämie
β-Thalassämie, homozygot ● mit leichter β+-Thalassämie ● gleichzeitig α-Thalassämie ● gesteigerte Fähigkeit zur fetalen
Hämoglobinproduktion (γ-Globin)
Heterozygote β0-Thalassämie β+-Thalassämie mit erblicher Persistenz
des fetalen Hämoglobins
δβ-Thalassämie
β-Thalassämie, heterozygot ● gleichzeitig Vererbung zusätzlicher
α-Gene ● dominante β-Thalassämie-Anlage
α0-Thalassämie α+-Thalassämie
δβ-Thalassämie und erbliche Persistenz des fetalen Hämoglobins ● homozygote δβ-Thalassämie ● heterozygote δβ-Thalassämie/β-Thalassämie ● homozygotes Lepore-Hb (fusionierte δβ-Ketten)
Hämoglobin-H-Krankheit (4β-Ketten)
krozytärer geringgradiger Anämie, die keiner Therapie bedarf.
Deren klare Diagnose hat grosse Bedeutung für die genetische
Beratung von merkmaltragenden Eltern.
■
PD Dr. med. Holger N. Lode Otto-Heubner-Zentrum für Kinder- und Jugendmedizin
Charité Universitätsmedizin Berlin, CVK Augustenburger Platz 1 D-13353 Berlin
E-Mail: holger.lode@charite.de
Interessenkonflikte: keine deklariert
Diese Arbeit erschien zuerst in «Notfall & Hausarztmedizin» 2006; 32 (12). Die Übernahme erfolgt mit freundlicher Genehmigung von Verlag und Autor.
Literatur: 1. Gadner H, Gaedicke G, Niemeyer C, Ritter J: Pädiatrische Hämatologie und Onkologie. Heidelberg: Springer,
2005. 2. Farhi DC, Luebbers EL, Rosenthal NS: Bone marrow biopsy findings in childhood anemia: prevalence of transient
erythroblastopenia of childhood. Arch Pathol Lab Med 1998; 122: 638—641. 3. Kynaston JA, West NC, Reid MM: A regional experienceof red cell aplasia. Eur J Pediatr 1993; 152: 306—308. 4. Skeppner G, Wranne L: Transient erythroblastopenia of childhood in Sweden: incidence and findings at the time
of diagnosis. Acta Paediatr 1993; 82: 574—578. 5. Cherrick I, Karayalcin G, Lanzkowsky R: Transient erythroblastopenia of childhood. Prospective study of fifty
patients. Am J Pediatr Hematol Oncol 1994; 16: 320—324. 6. Chan GC, Kanwar VS, Wilimas J: Transient erythroblastopenia of childhood associated with transient neurologic
deficit: report of a case and review of the literature. J Paediatr Child Health 1998; 34: 299—301. 7. Michelson AD, Marshall PC: Transient neurological disorder associated with transient erythroblastopenia of
childhood. Am J Pediatr Hematol Oncol 1987; 9: 161—163. 8. Willig TN, Niemeyer CM, Leblanc T et al.: Identification of new prognosis factors from the clinical and epide-
miologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Société d'Hématologie et d'Immunologie Pédiatrique (SHIP), Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI). Pediatr Res 1999; 46: 553—561. 9. Willig TN, Perignon JL,Gustavsson P et al.: High adenosine deaminase level among healthy probands of Diamond-Blackfan anemia (DBA) cosegregates with the DBA gene region on chromosome 19q13. The DBA Working Group of Societe d'Immunologie Pediatrique (SHIP). Blood 1998; 92: 4422—4427. 10. CATHIE IA: Erythrogenesis imperfecta. Arch Dis Child 1950; 25: 313—324. 11. Lipton JM, Atsidaftos E, Zyskind I, Vlachos A: Improving clinical care and elucidating the pathophysiology of Diamond-Blackfan anemia: an update from the Diamond-Blackfan Anemia Registry. Pediatr Blood Cancer 2006; 46: 558—564. 12. Vlachos A, Klein GW, Lipton JM: The Diamond-Blackfan Anemia Registry: tool for investigating the epidemiology and biology of Diamond-Blackfan anemia. J Pediatr Hematol Oncol 2001: 23: 377—382. 13. Willig TN, Draptchinskaia N, Dianzani I et al.: Mutations in ribosomal protein S19 gene and diamond blackfan anemia: wide variations in phenotypic expression. Blood 1999: 94: 4294—4306. 14. Proust A, Da CL, Rince P et al.: Ten novel Diamond-Blackfan anemia mutations and three polymorphisms within the rps19 gene. Hematol J 2003: 4: 132—136. 15. Gazda H, Lipton JM, Willig TN et al.: Evidence for linkage of familial Diamond-Blackfan anemia to chromosome 8p23.3-p22 and for non-19q non-8p disease. Blood 2001; 97: 2145—2150. 16. Wickramasinghe SN: Diagnosis of megaloblastic anaemias. Blood Rev 2006. 17. Irwin JJ, Kirchner JT: Anemia in children. Am Fam Physician 2001; 64: 1379—1386. 18. Mentzer WC Jr. Differentiation of iron deficiency from thalassaemia trait. Lancet 1973; 1: 882. 19. Demir A, Yarali N, Fisgin T, Duru F, Kara A: Most reliable indices in differentlation between thalassemia trait and iron deficiency anemia. Pediatr Int 2002; 44: 612—616. 20. Hoffbrand AV, Pettit JE, Moss PAH, Hoelzer D: Grundkurs Hämatologie. Berlin, Wien: Blackwell WissenschaftsVerlag, 2003.
674 ARS MEDICI 15 ■ 2008