
Transkript
FORUM
Neue Standards – höhere Risiken?
Die Leitlinien für Good Distribution Practice (GDP) präsentieren sich im hohen Alter von gegen 20 Jahren nicht mehr taufrisch – in der Zwischenzeit haben sich die Anforderungen bezüglich Patientensicherheit beträchtlich verändert. Auch weil immer mehr aufwendig hergestellte Medikamente auf den Markt kommen. Was ist zu erwarten? Erste Einschätzungen.
HANS WIRZ
Wenn vom Staat neue Regelungen kommen, steigen meistens die Kosten. Bei wem? In diesem Fall beim Grosshandel, was zu Mehrkosten bei der Umsetzung der neuen Vorschriften bei den Apotheken, Spitälern und SD-Ärzten führen wird. Auslöser für neue Vorschriften ist die Richtlinie 2011/62 der
EU. Dass die erstmals 1994 in Kraft getretenen GDP-Leitlinien durch neue abgelöst werden sollen, erstaunt angesichts der rasanten Entwicklungen bei den Arzneimitteln niemand. Stichworte dazu sind Biologicals und Biosimilars als Nachfolger von «gewöhnlichen» Medikamenten und Generika.
Der Mechanismus und die neue Regelung Wenn es die Europäische Zulassungsbehörde (EMA) für angebracht hält, erarbeitet sie im Rahmen der EU-Kommission neue Leitlinien. Diese gehen dann für eine Periode von sechs Monaten in den Mitgliederländern der EU in die Vernehmlassung, um schliesslich in definitiver Form – Commission Guidelines on Good Distribution Practice of Medicinal Products for Human Use – neue Vorschriften vorzugeben. GIRP, der Europäische Verband der pharmazeutischen Grosshändler, hat das Thema in einer Medienmitteilung vom 4. Juni 2012 aufgegriffen.
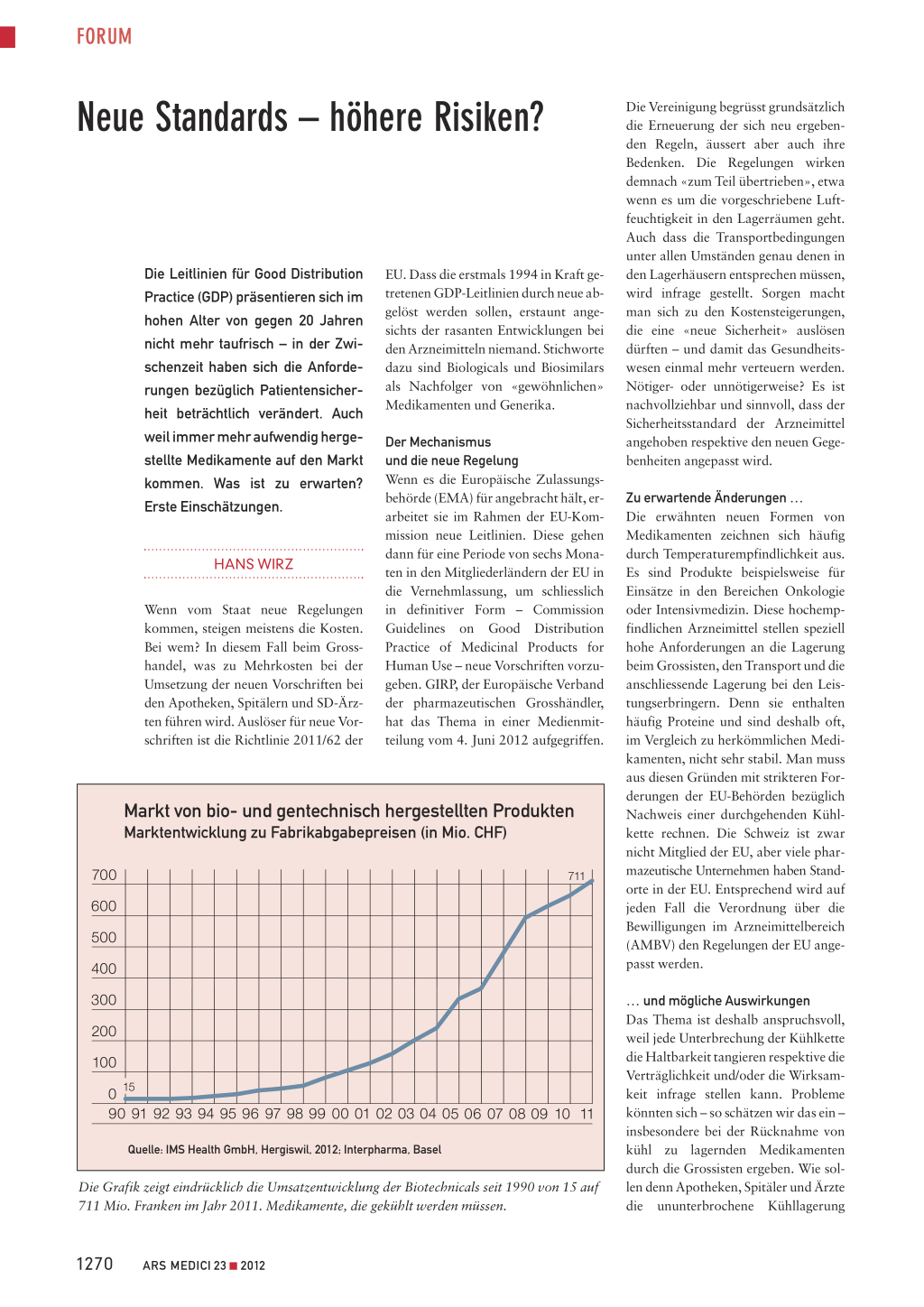
Markt von bio- und gentechnisch hergestellten Produkten
Marktentwicklung zu Fabrikabgabepreisen (in Mio. CHF)
700 711 600 500 400
Die Vereinigung begrüsst grundsätzlich die Erneuerung der sich neu ergebenden Regeln, äussert aber auch ihre Bedenken. Die Regelungen wirken demnach «zum Teil übertrieben», etwa wenn es um die vorgeschriebene Luftfeuchtigkeit in den Lagerräumen geht. Auch dass die Transportbedingungen unter allen Umständen genau denen in den Lagerhäusern entsprechen müssen, wird infrage gestellt. Sorgen macht man sich zu den Kostensteigerungen, die eine «neue Sicherheit» auslösen dürften – und damit das Gesundheitswesen einmal mehr verteuern werden. Nötiger- oder unnötigerweise? Es ist nachvollziehbar und sinnvoll, dass der Sicherheitsstandard der Arzneimittel angehoben respektive den neuen Gegebenheiten angepasst wird.
Zu erwartende Änderungen … Die erwähnten neuen Formen von Medikamenten zeichnen sich häufig durch Temperaturempfindlichkeit aus. Es sind Produkte beispielsweise für Einsätze in den Bereichen Onkologie oder Intensivmedizin. Diese hochempfindlichen Arzneimittel stellen speziell hohe Anforderungen an die Lagerung beim Grossisten, den Transport und die anschliessende Lagerung bei den Leistungserbringern. Denn sie enthalten häufig Proteine und sind deshalb oft, im Vergleich zu herkömmlichen Medikamenten, nicht sehr stabil. Man muss aus diesen Gründen mit strikteren Forderungen der EU-Behörden bezüglich Nachweis einer durchgehenden Kühlkette rechnen. Die Schweiz ist zwar nicht Mitglied der EU, aber viele pharmazeutische Unternehmen haben Standorte in der EU. Entsprechend wird auf jeden Fall die Verordnung über die Bewilligungen im Arzneimittelbereich (AMBV) den Regelungen der EU angepasst werden.
300 200 100
0 15 90 91 92 93 94 95 96 97 98 99 00 01 02 03 04 05 06 07 08 09 10 11
Quelle: IMS Health GmbH, Hergiswil, 2012; Interpharma, Basel
Die Grafik zeigt eindrücklich die Umsatzentwicklung der Biotechnicals seit 1990 von 15 auf 711 Mio. Franken im Jahr 2011. Medikamente, die gekühlt werden müssen.
… und mögliche Auswirkungen Das Thema ist deshalb anspruchsvoll, weil jede Unterbrechung der Kühlkette die Haltbarkeit tangieren respektive die Verträglichkeit und/oder die Wirksamkeit infrage stellen kann. Probleme könnten sich – so schätzen wir das ein – insbesondere bei der Rücknahme von kühl zu lagernden Medikamenten durch die Grossisten ergeben. Wie sollen denn Apotheken, Spitäler und Ärzte die ununterbrochene Kühllagerung
1270 ARS MEDICI 23 ■ 2012
belegen? Bis jetzt ist es so, dass die
Abnehmer bei Nichtgebrauch der
Medikamente, etwa wegen Falsch-
bestellungen, Therapiewechsel oder
Falschlieferungen, diese problemlos
zurückschicken konnten. Bei den Gros-
sisten wurden sie dann vermutlich je
nach fachlicher Beurteilung fachge-
recht weiter gelagert und bei der nächs-
ten Gelegenheit wieder ausgeliefert.
Wir zweifeln daran, dass das weiterhin
möglich sein wird. Und: Wer trägt im
Einzelfall die durch die neue GDP ent-
stehenden Kosten? Die Patienten, die
verschreibenden Ärzte, der Grosshan-
del – oder die Apotheken? Und wie
kann die Patientensicherheit gewähr-
leistet werden?
In der Schweiz werden vom Grosshan-
del pro Tag schätzungsweise 600 000
Packungen Arzneimittel ausgeliefert.
Uns scheint die Kühlkettenproblematik
naheliegend, aber es können sich natür-
lich aus der neuen GDP auch andere
Auswirkungen ergeben, die im Moment
noch nicht zu erkennen sind. GIRP er-
wartet die Publikation der neuen GDP-
Leitlinien im Spätherbst 2012; die Um-
setzungsfrist wird dann sechs Monate
betragen.
❖
Hans Wirz, Redaktor OTX World Erstpublikation in OTX World Nr. 86, November 2012.